

Identification of a novel cell type-specific intronic enhancer of macrophage migration inhibitory factor (MIF) and its regulation by mithramycin
- PMID: 21087445
- PMCID: PMC3043308
- DOI: 10.1111/j.1365-2249.2010.04289.x
Identification of a novel cell type-specific intronic enhancer of macrophage migration inhibitory factor (MIF) and its regulation by mithramycin
Abstract
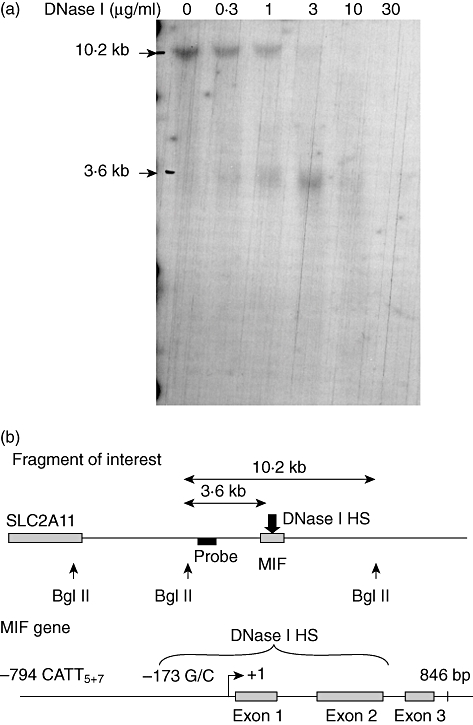
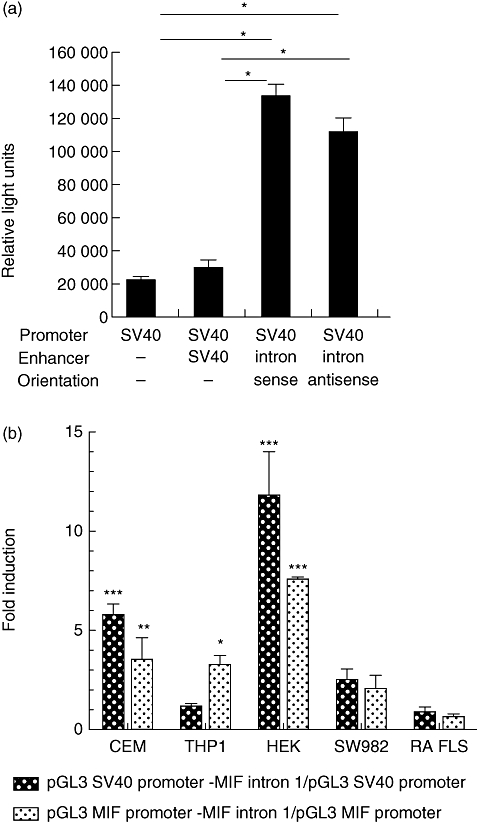
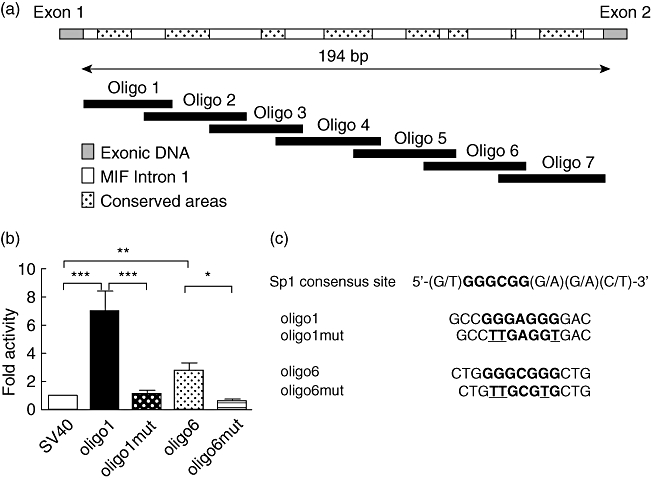
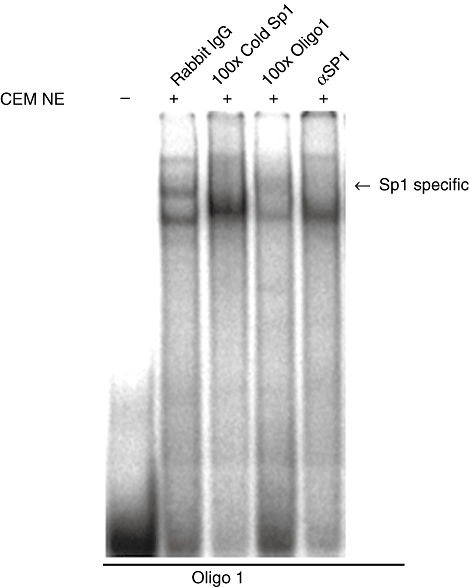
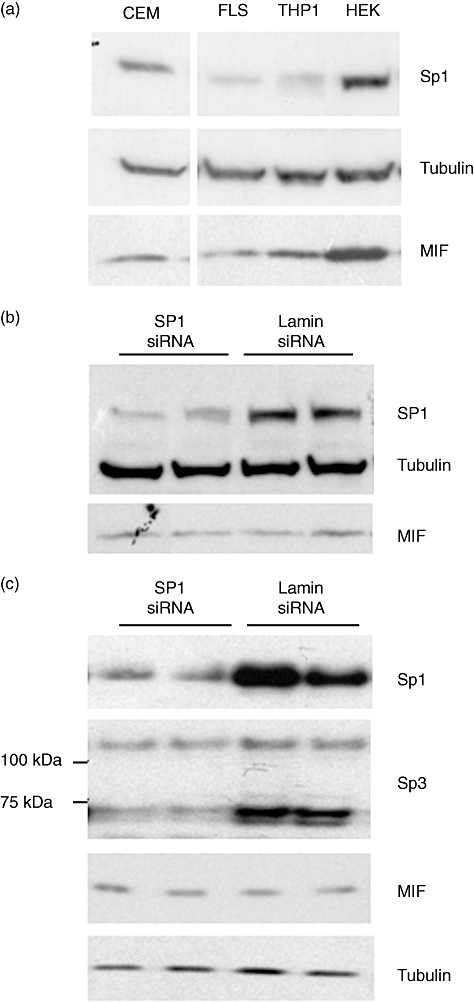
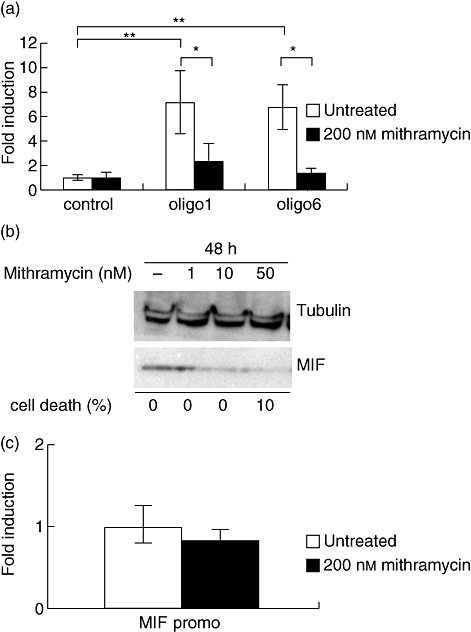
The aim of this study was to determine the genetic regulation of macrophage migration inhibitory factor (MIF). DNase I hypersensitivity was used to identify potential hypersensitive sites (HS) across the MIF gene locus. Reporter gene assays were performed in different human cell lines with constructs containing the native or mutated HS element. Following phylogenetic and transcription factor binding profiling, electrophoretic mobility shift assay (EMSA) and RNA interference were performed and the effects of incubation with mithramycin, an antibiotic that binds GC boxes, were also studied. An HS centred on the first intron of MIF was identified. The HS acted as an enhancer in human T lymphoblasts (CEMC7A), human embryonic kidney cells (HEK293T) and human monocytic cells (THP-1), but not in a fibroblast-like synoviocyte (FLS) cell line (SW982) or cultured FLS derived from rheumatoid arthritis (RA) patients. Two cis-elements within the first intron were found to be responsible for the enhancer activity. Mutation of the consensus Sp1 GC box on each cis-element abrogated enhancer activity and EMSA indicated Sp1 binding to one of the cis-elements contained in the intron. SiRNA knock-down of Sp1 alone or Sp1 and Sp3 together was incomplete and did not alter the enhancer activity. Mithramycin inhibited expression of MIF in CEMC7A cells. This effect was specific to the intronic enhancer and was not seen on the MIF promoter. These results identify a novel, cell type-specific enhancer of MIF. The enhancer appears to be driven by Sp1 or related Sp family members and is highly sensitive to inhibition via mithramycin.
© 2010 The Authors. Clinical and Experimental Immunology © 2010 British Society for Immunology.
Figures
References
-
- Bernhagen J, Calandra T, Mitchell RA, et al. MIF is a pituitary-derived cytokine that potentiates lethal endotoxaemia. Nature. 1993;365:756–9. - PubMed
-
- Kudrin A, Scott M, Martin S, et al. Human macrophage migration inhibitory factor: a proven immunomodulatory cytokine. J Biol Chem. 2006;281:29641–51. - PubMed
-
- Mikulowska A, Metz CN, Bucala R, Holmdahl R. Macrophage migration inhibitory factor is involved in the pathogenesis of collagen type II-induced arthritis in mice. J Immunol. 1997;158:5514–17. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
