

Predicting genome-wide redundancy using machine learning
- PMID: 21087504
- PMCID: PMC2998534
- DOI: 10.1186/1471-2148-10-357
Predicting genome-wide redundancy using machine learning
Abstract
Background: Gene duplication can lead to genetic redundancy, which masks the function of mutated genes in genetic analyses. Methods to increase sensitivity in identifying genetic redundancy can improve the efficiency of reverse genetics and lend insights into the evolutionary outcomes of gene duplication. Machine learning techniques are well suited to classifying gene family members into redundant and non-redundant gene pairs in model species where sufficient genetic and genomic data is available, such as Arabidopsis thaliana, the test case used here.
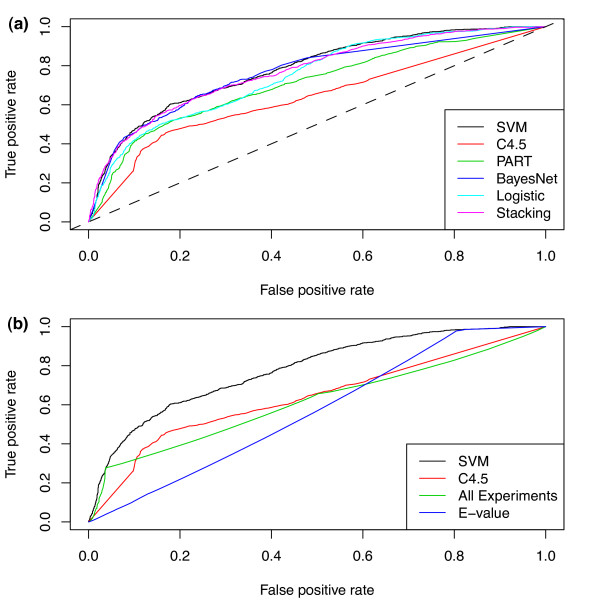
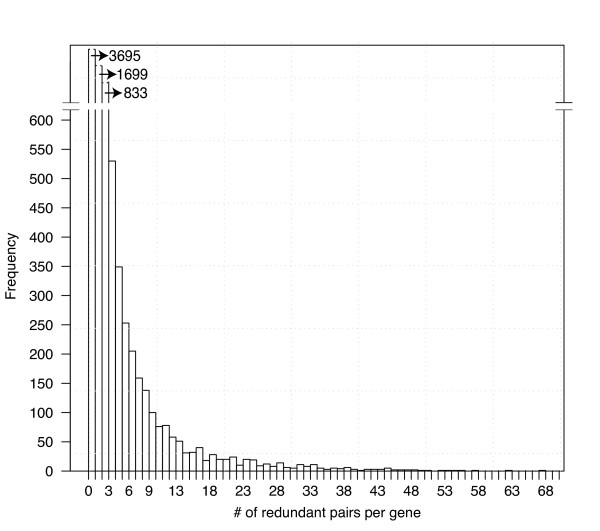
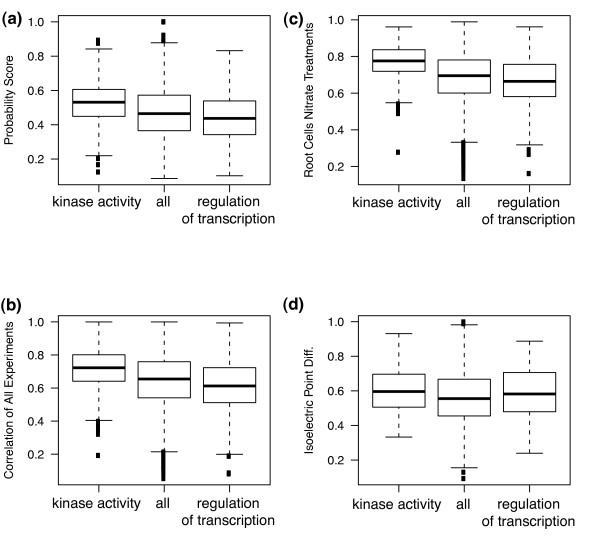
Results: Machine learning techniques that combine multiple attributes led to a dramatic improvement in predicting genetic redundancy over single trait classifiers alone, such as BLAST E-values or expression correlation. In withholding analysis, one of the methods used here, Support Vector Machines, was two-fold more precise than single attribute classifiers, reaching a level where the majority of redundant calls were correctly labeled. Using this higher confidence in identifying redundancy, machine learning predicts that about half of all genes in Arabidopsis showed the signature of predicted redundancy with at least one but typically less than three other family members. Interestingly, a large proportion of predicted redundant gene pairs were relatively old duplications (e.g., Ks > 1), suggesting that redundancy is stable over long evolutionary periods.
Conclusions: Machine learning predicts that most genes will have a functionally redundant paralog but will exhibit redundancy with relatively few genes within a family. The predictions and gene pair attributes for Arabidopsis provide a new resource for research in genetics and genome evolution. These techniques can now be applied to other organisms.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
