

Inhibitors of type 5 17β-hydroxysteroid dehydrogenase (AKR1C3): overview and structural insights
- PMID: 21087665
- PMCID: PMC3047600
- DOI: 10.1016/j.jsbmb.2010.11.004
Inhibitors of type 5 17β-hydroxysteroid dehydrogenase (AKR1C3): overview and structural insights
Abstract
There is considerable interest in the development of an inhibitor of aldo-keto reductase (AKR) 1C3 (type 5 17β-hydroxysteroid dehydrogenase and prostaglandin F synthase) as a potential therapeutic for both hormone-dependent and hormone-independent cancers. AKR1C3 catalyzes the reduction of 4-androstene-3,17-dione to testosterone and estrone to 17β-estradiol in target tissues, which will promote the proliferation of hormone dependent prostate and breast cancers, respectively. AKR1C3 also catalyzes the reduction of prostaglandin (PG) H(2) to PGF(2α) and PGD(2) to 9α,11β-PGF(2), which will limit the formation of anti-proliferative prostaglandins, including 15-deoxy-Δ(12,14)-PGJ(2), and contribute to proliferative signaling. AKR1C3 is overexpressed in a wide variety of cancers, including breast and prostate cancer. An inhibitor of AKR1C3 should not inhibit the closely related isoforms AKR1C1 and AKR1C2, as they are involved in other key steroid hormone biotransformations in target tissues. Several structural leads have been explored as inhibitors of AKR1C3, including non-steroidal anti-inflammatory drugs, steroid hormone analogues, flavonoids, cyclopentanes, and benzodiazepines. Inspection of the available crystal structures of AKR1C3 with multiple ligands bound, along with the crystal structures of the other AKR1C isoforms, provides a structural basis for the rational design of isoform specific inhibitors of AKR1C3. We find that there are subpockets involved in ligand binding that are considerably different in AKR1C3 relative to the closely related AKR1C1 or AKR1C2 isoforms. These pockets can be used to further improve the binding affinity and selectivity of the currently available AKR1C3 inhibitors. Article from the special issue on Targeted Inhibitors.
Copyright © 2010 Elsevier Ltd. All rights reserved.
Figures
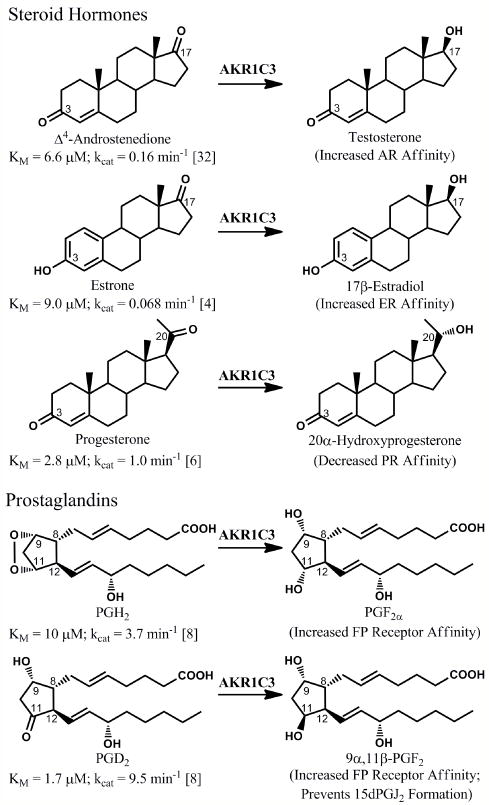
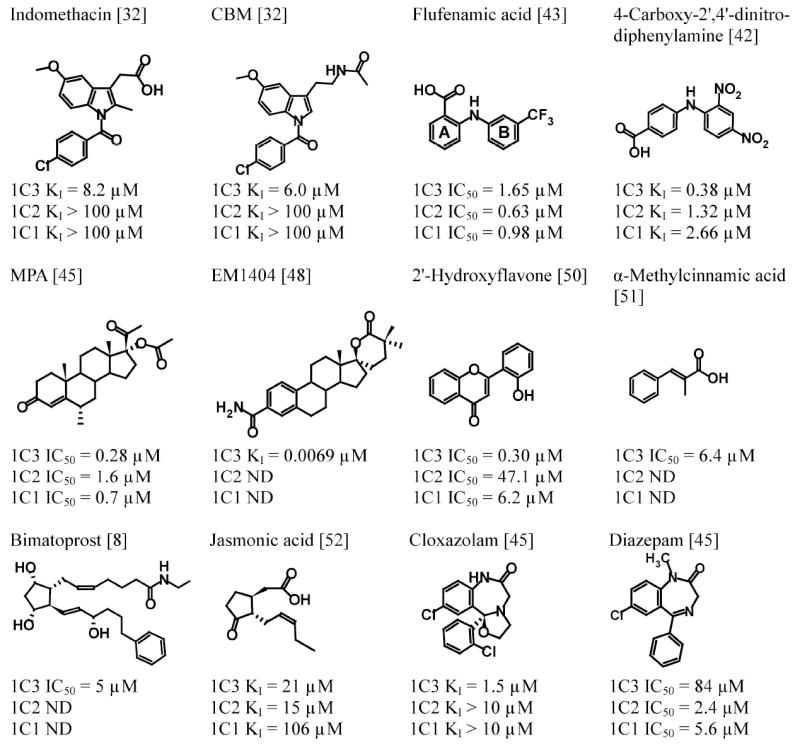
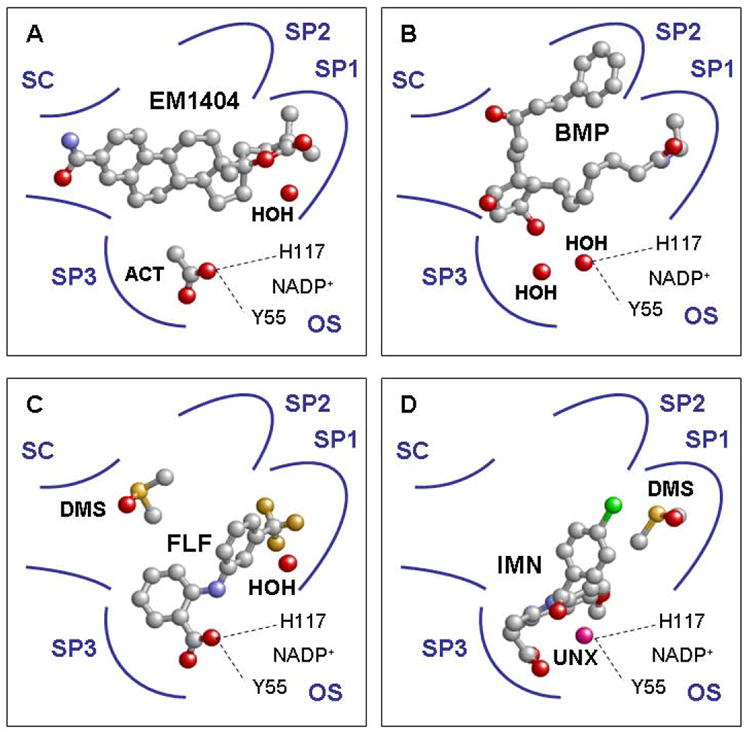
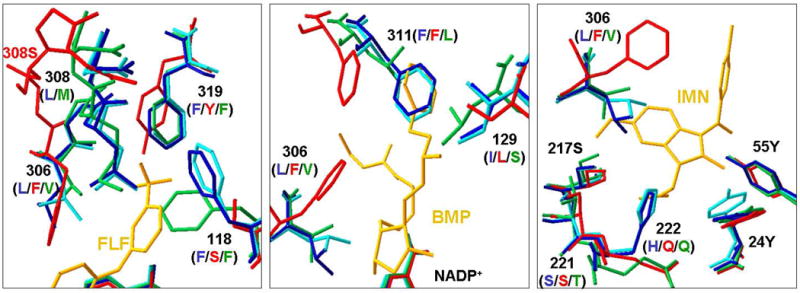
References
-
- Marchais-Oberwinkler S, Hennl C, Möller G, Klein T, Negri M, Oster A, Spadaro A, Werth R, Wetzel M, Xu K, Frotscher M, Hartmann RW, Adamski J. 17β-Hydroxysteroid dehydrogenase (17β-HSD): gene, protein structures, novel therapeutic targets and recent progress in inhibitor development. J Steroid Biochem Mol Biol. 2010 current issue. - PubMed
-
- Poirier D. Contribution to the development of inhibitors of 17β-hydroxysteroid dehydrogenase types 1 and 7: key tools for studying and treating estrogen-dependent diseases. J Steroid Biochem Mol Biol. 2010 current issue. - PubMed
-
- Dufort I, Rheault P, Huang X-F, Soucy P, Luu-The V. Characteristics of a highly labile human type 5 17β-hydroxysteroid dehydrogenase. Endocrinology. 1999;140:568–574. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
