

Anticancer activity of Celastrol in combination with ErbB2-targeted therapeutics for treatment of ErbB2-overexpressing breast cancers
- PMID: 21088503
- PMCID: PMC3047084
- DOI: 10.4161/cbt.11.2.13959
Anticancer activity of Celastrol in combination with ErbB2-targeted therapeutics for treatment of ErbB2-overexpressing breast cancers
Abstract
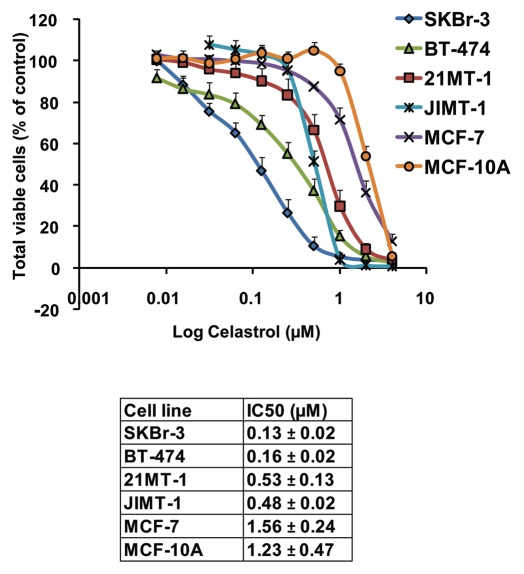
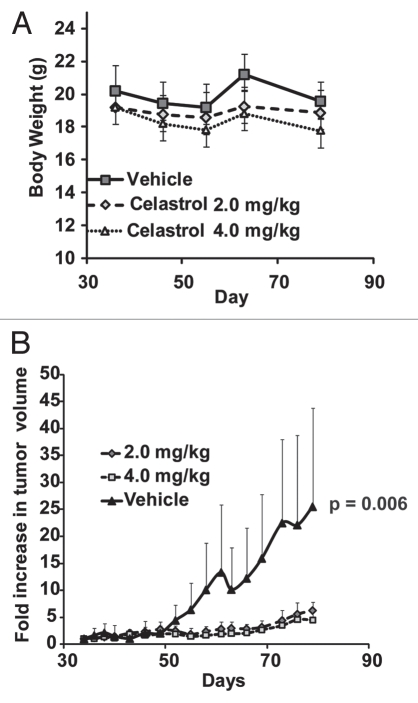
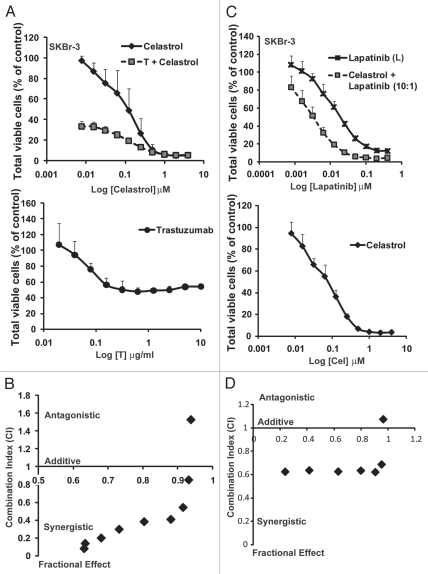
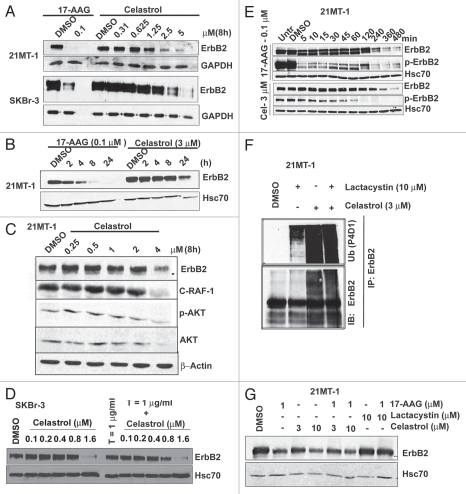
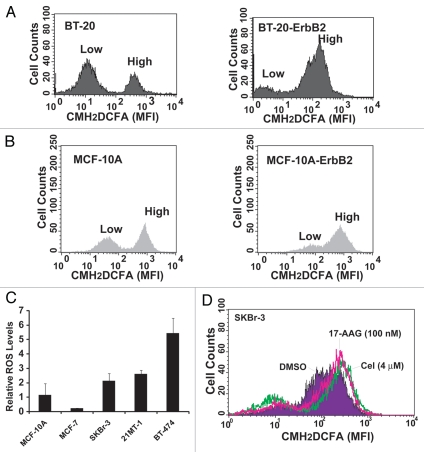
The receptor tyrosine kinase ErbB2 is overexpressed in up to a third of breast cancers, allowing targeted therapy with ErbB2-directed humanized antibodies such as Trastuzumab. Concurrent targeting of ErbB2 stability with HSP90 inhibitors is synergistic with Trastuzumab, suggesting that pharmacological agents that can inhibit HSP90 as well as signaling pathways activated by ErbB2 could be useful against ErbB2-overexpressing breast cancers. The triterpene natural product Celastrol inhibits HSP90 and several pathways relevant to ErbB2-dependent oncogenesis including the NFκB pathway and the proteasome, and has shown promising activity in other cancer models. Here, we demonstrate that Celastrol exhibits in vitro antitumor activity against a panel of human breast cancer cell lines with selectivity towards those overexpressing ErbB2. Celastrol strongly synergized with ErbB2-targeted therapeutics Trastuzumab and Lapatinib, producing higher cytotoxicity with substantially lower doses of Celastrol. Celastrol significantly retarded the rate of growth of ErbB2-overexpressing human breast cancer cells in a mouse xenograft model with only minor systemic toxicity. Mechanistically, Celastrol not only induced the expected ubiquitinylation and degradation of ErbB2 and other HSP90 client proteins, but it also increased the levels of reactive oxygen species (ROS). Our studies show that the Michael Acceptor functionality in Celastrol is important for its ability to destabilize ErbB2 and exert its bioactivity against ErbB2-overexpressing breast cancer cells. These studies suggest the potential use of Michael acceptor-containing molecules as novel therapeutic modalities against ErbB2-driven breast cancer by targeting multiple biological attributes of the driver oncogene.
Figures

Similar articles
-
Marked enhancement of lysosomal targeting and efficacy of ErbB2-targeted drug delivery by HSP90 inhibition.Oncotarget. 2016 Mar 1;7(9):10522-35. doi: 10.18632/oncotarget.7231. Oncotarget. 2016. PMID: 26859680 Free PMC article.
-
Antitumor efficacy of IPI-504, a selective heat shock protein 90 inhibitor against human epidermal growth factor receptor 2-positive human xenograft models as a single agent and in combination with trastuzumab or lapatinib.Mol Cancer Ther. 2009 Aug;8(8):2131-41. doi: 10.1158/1535-7163.MCT-08-1038. Epub 2009 Aug 11. Mol Cancer Ther. 2009. PMID: 19671750
-
Activity of the dual kinase inhibitor lapatinib (GW572016) against HER-2-overexpressing and trastuzumab-treated breast cancer cells.Cancer Res. 2006 Feb 1;66(3):1630-9. doi: 10.1158/0008-5472.CAN-05-1182. Cancer Res. 2006. PMID: 16452222
-
Acquired resistance to small molecule ErbB2 tyrosine kinase inhibitors.Clin Cancer Res. 2008 Nov 1;14(21):6730-4. doi: 10.1158/1078-0432.CCR-08-0581. Clin Cancer Res. 2008. PMID: 18980964 Review.
-
HER-2-positive breast cancer: hope beyond trastuzumab.BioDrugs. 2007;21(2):69-77. doi: 10.2165/00063030-200721020-00001. BioDrugs. 2007. PMID: 17402790 Review.
Cited by
-
Polypeptide-based nanogels co-encapsulating a synergistic combination of doxorubicin with 17-AAG show potent anti-tumor activity in ErbB2-driven breast cancer models.J Control Release. 2015 Jun 28;208:59-66. doi: 10.1016/j.jconrel.2015.02.001. Epub 2015 Feb 3. J Control Release. 2015. PMID: 25660204 Free PMC article.
-
Marked enhancement of lysosomal targeting and efficacy of ErbB2-targeted drug delivery by HSP90 inhibition.Oncotarget. 2016 Mar 1;7(9):10522-35. doi: 10.18632/oncotarget.7231. Oncotarget. 2016. PMID: 26859680 Free PMC article.
-
Mechanisms of Trastuzumab resistance in ErbB2-driven breast cancer and newer opportunities to overcome therapy resistance.J Carcinog. 2011;10:28. doi: 10.4103/1477-3163.90442. Epub 2011 Nov 30. J Carcinog. 2011. PMID: 22190870 Free PMC article.
-
Cytoplasmic Vacuolization: A Fascinating Morphological Alteration From Cellular Stress to Cell Death.Cancer Sci. 2025 May;116(5):1181-1192. doi: 10.1111/cas.70013. Epub 2025 Feb 27. Cancer Sci. 2025. PMID: 40017124 Free PMC article. Review.
-
Synergistic anticancer effects of triptolide and celastrol, two main compounds from thunder god vine.Oncotarget. 2015 Oct 20;6(32):32790-804. doi: 10.18632/oncotarget.5411. Oncotarget. 2015. PMID: 26447544 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
- CA105489/CA/NCI NIH HHS/United States
- T32 CA 009476/CA/NCI NIH HHS/United States
- R01 CA116552/CA/NCI NIH HHS/United States
- R01 CA105489/CA/NCI NIH HHS/United States
- CA94143/CA/NCI NIH HHS/United States
- P30 CA036727/CA/NCI NIH HHS/United States
- CA127239/CA/NCI NIH HHS/United States
- CA99163/CA/NCI NIH HHS/United States
- R01 CA099163/CA/NCI NIH HHS/United States
- T32 CA009476/CA/NCI NIH HHS/United States
- R01 CA127239/CA/NCI NIH HHS/United States
- CA87986/CA/NCI NIH HHS/United States
- R01 CA087986/CA/NCI NIH HHS/United States
- 5P30 CA036727/CA/NCI NIH HHS/United States
- R01 CA096844/CA/NCI NIH HHS/United States
- CA96844/CA/NCI NIH HHS/United States
- R01 CA094143/CA/NCI NIH HHS/United States
- CA116552/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous