

Review
doi: 10.1038/nri2873.
Epub 2010 Nov 19.
Sterile inflammation: sensing and reacting to damage
Affiliations
- PMID: 21088683
- PMCID: PMC3114424
- DOI: 10.1038/nri2873
Item in Clipboard
Review
Sterile inflammation: sensing and reacting to damage
Nat Rev Immunol.
2010 Dec.
Abstract
Over the past several decades, much has been revealed about the nature of the host innate immune response to microorganisms, with the identification of pattern recognition receptors (PRRs) and pathogen-associated molecular patterns, which are the conserved microbial motifs sensed by these receptors. It is now apparent that these same PRRs can also be activated by non-microbial signals, many of which are considered as damage-associated molecular patterns. The sterile inflammation that ensues either resolves the initial insult or leads to disease. Here, we review the triggers and receptor pathways that result in sterile inflammation and its impact on human health.
Figures
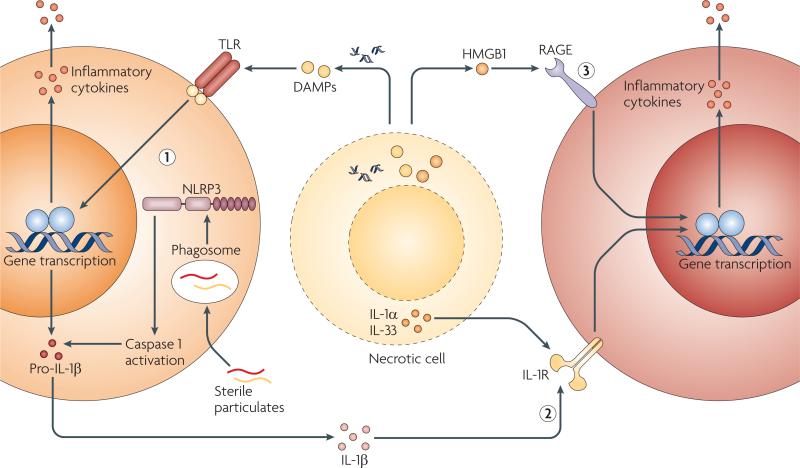
Sterile stimuli that include damage-associated molecular patterns (DAMPs), sterile particulates and intracellular cytokines released from necrotic cells can activate the host immune system to induce sterile inflammation through at least three pathways that are not mutually exclusive. DAMPs and sterile particulates can active host pathogen recognition receptors (PRRs), such as the Toll-like receptors (TLRs) and the nucleotide-binding oligomerization domain (NOD)-like receptor NLRP3 (NOD-, LRR- and pyrin domain-containing 3), which are also used by the host to sense microorganisms. Activation of these receptors results in the upregulation of cytokines and chemokines, such as interleukin-1β (IL-1β), which are released to recruit and activate additional inflammatory cells (1). Intracellular cytokines such as IL-1α and IL-33 that are released by damaged, necrotic cells activate signalling pathways downstream of PRRs (2). Endogenous DAMPs signal directly through host receptors that are not typically considered to be PRRs or to be involved in microbial detection (3). HMGB1, high-mobility group box 1; IL-1R, IL-1 receptor; RAGE, receptor for advanced glycation end-products.

The activation of the NLRP3 (NOD-, LRR- and pyrin domain-containing 3) inflammasome has been associated with three separate phenomena. ATP can bind to purinergic receptor P2X7 (P2RX7), which then opens a cation channel and a large pore through pannexin 1 that, in turn, can lead to ionic fluxes, including intracellular K+ depletion, and other events that are poorly understood. Endocytosis of sterile particulates, such as silica, asbestos and cholesterol crystals, results in lysosomal damage and membrane destabilization, leading to activation of the protease cathepsin B. The generation of reactive oxygen species (ROS) during cellular stress or death has been associated with NLRP3 activation, although the role of ROS in NLRP3 activation remains controversial. DAMPs, damage-associated molecular patterns; IL-1β, interleukin-1β.

Necrotic cells release the precursor form of interleukin-1α (IL-1α), which is biologically active and stimulates neighbouring parenchymal cells, through IL-1 receptor (IL-1R), to secrete the chemokine CXC-chemokine ligand 1 (CXCL1). In addition, IL-1α can stimulate resident macrophages to produce additional IL-1α and IL-1β through a caspase 1-independent mechanism that further boosts CXCL1 secretion. In turn, CXCL1 functions through CXC-chemokine receptor 2 (CXCR2) on neutrophils to recruit them to the site of injury. This figure is based on published studies using a peritoneal model of sterile inflammation,,.
Comment in
-
Heat shock proteins are no DAMPs, rather 'DAMPERs'.Nat Rev Immunol. 2011 Jul 25;11(8):565; author reply 565. doi: 10.1038/nri2873-c1. Nat Rev Immunol. 2011. PMID: 21785457 No abstract available.
References
-
- Mossman BT, Churg A. Mechanisms in the pathogenesis of asbestosis and silicosis. Am. J. Respir. Crit. Care Med. 1998;157:1666–1680. - PubMed
-
- Cotran RS, Kumar V, Robbins S. In: Robbins Pathologic Basis of Disease. Schoen FJ, editor. W. B. Saunders Company; Philadelphia: 1994. pp. 6–11.
-
- Cotran RS, Kumar V, Robbins S. In: Robbins Pathologic Basis of Disease. Schoen FJ, editor. W. B. Saunders Company; Philadelphia: 1994. pp. 1255–1259.
-
- Weiner HL, Frenkel D. Immunology and immunotherapy of Alzheimer's disease. Nature Rev. Immunol. 2006;6:404–416. - PubMed
-
- Ross R. Atherosclerosis — an inflammatory disease. N. Engl. J. Med. 1999;340:115–126. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
