

The paradox of the neutrophil's role in tissue injury
- PMID: 21097697
- PMCID: PMC6608002
- DOI: 10.1189/jlb.0910538
The paradox of the neutrophil's role in tissue injury
Abstract
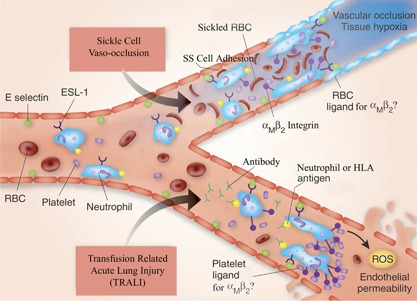
The neutrophil is an essential component of the innate immune system, and its function is vital to human life. Its production increases in response to virtually all forms of inflammation, and subsequently, it can accumulate in blood and tissue to varying degrees. Although its participation in the inflammatory response is often salutary by nature of its normal interaction with vascular endothelium and its capability to enter tissues and respond to chemotactic gradients and to phagocytize and kill microrganisms, it can contribute to processes that impair vascular integrity and blood flow. The mechanisms that the neutrophil uses to kill microorganisms also have the potential to injure normal tissue under special circumstances. Its paradoxical role in the pathophysiology of disease is particularly, but not exclusively, notable in seven circumstances: 1) diabetic retinopathy, 2) sickle cell disease, 3) TRALI, 4) ARDS, 5) renal microvasculopathy, 6) stroke, and 7) acute coronary artery syndrome. The activated neutrophil's capability to become adhesive to endothelium, to generate highly ROS, and to secrete proteases gives it the potential to induce local vascular and tissue injury. In this review, we summarize the evidence for its role as a mediator of tissue injury in these seven conditions, making it or its products potential therapeutic targets.
Figures
References
-
- Silliman, C. C. , McLaughlin, N. J. (2006) Transfusion‐related acute lung injury. Blood Rev. 20, 139–159. - PubMed
-
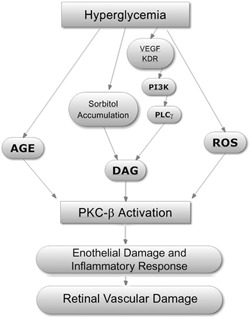
- Setter, S. M. , Campbell, R. K. , Cahoon, C. J. (2003) Biochemical pathways for microvascular complications of diabetes mellitus. Ann. Pharmacother. 37, 1858–1866. - PubMed
-
- Liu, X. J. , He, A. B. , Chang, Y. S. , Fang, F. D. (2006) Atypical protein kinase C in glucose metabolism. Cell. Signal. 18, 2071–2076. - PubMed
-
- Das Evcimen, N. , King, G. L. (2007) The role of protein kinase C activation and the vascular complications of diabetes. Pharmacol. Res. 55, 498–510. - PubMed
-
- Bloomgarden, Z. T. (2002) The epidemiology of complications. Diabetes Care 25, 924–932. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
