

BRAF gene amplification can promote acquired resistance to MEK inhibitors in cancer cells harboring the BRAF V600E mutation
- PMID: 21098728
- PMCID: PMC3372405
- DOI: 10.1126/scisignal.2001148
BRAF gene amplification can promote acquired resistance to MEK inhibitors in cancer cells harboring the BRAF V600E mutation
Abstract
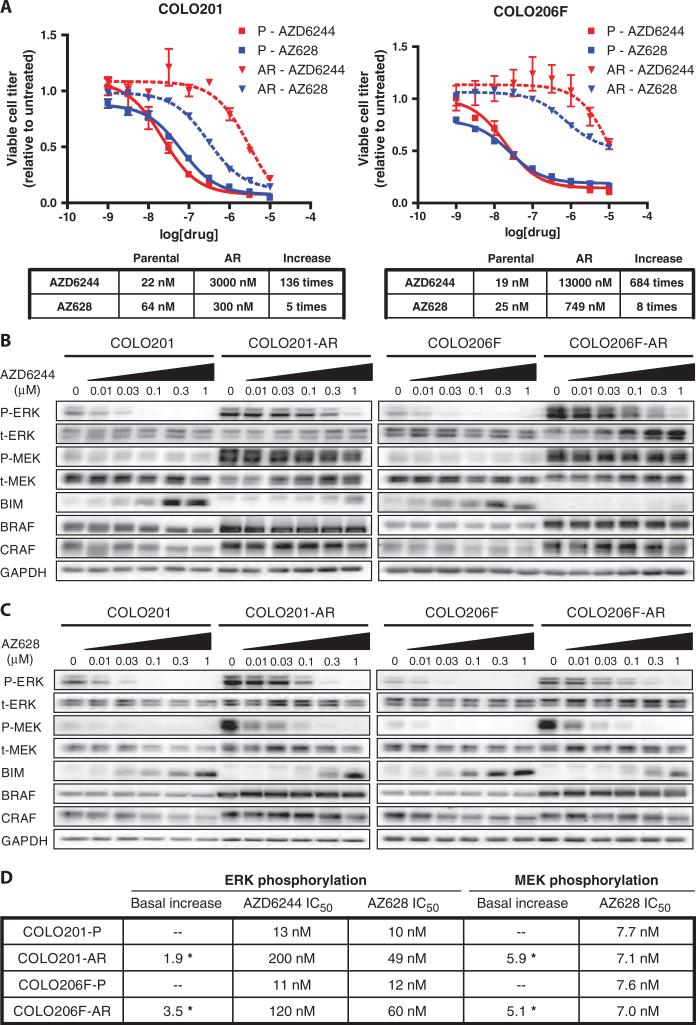
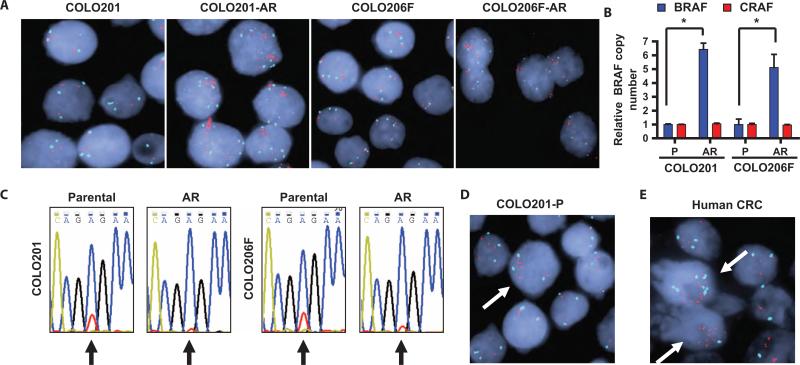
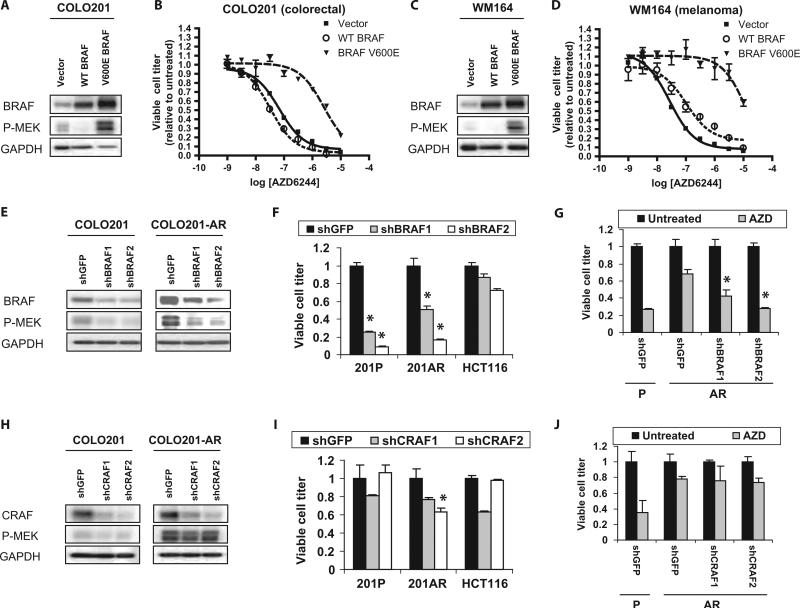
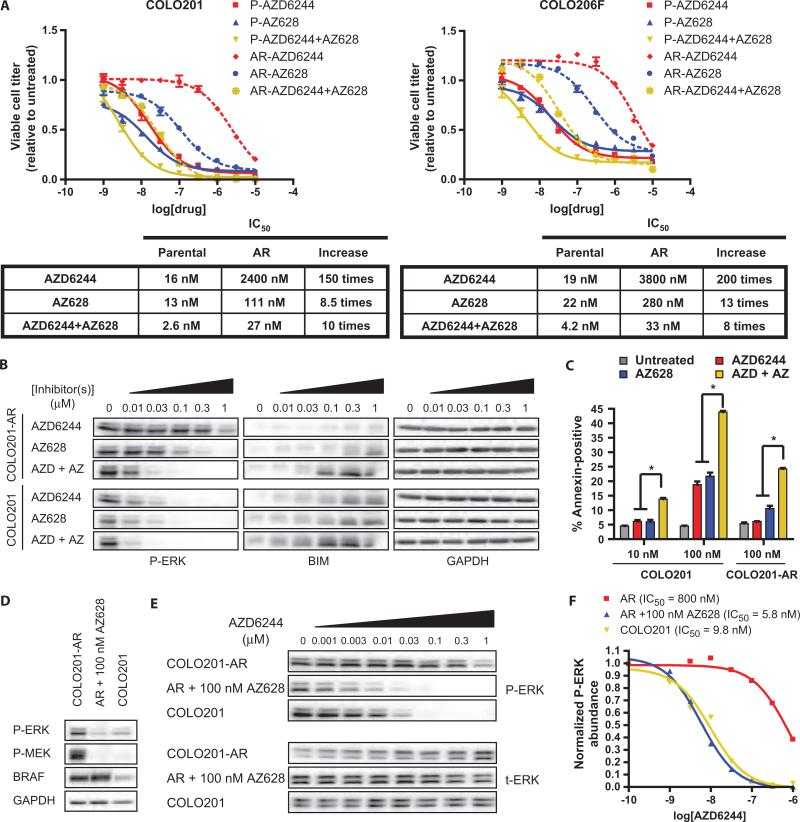
Oncogenic BRAF mutations are found in several tumor types, including melanomas and colorectal cancers. Tumors with BRAF mutations have increased mitogen-activated protein kinase pathway activity and heightened sensitivity to BRAF and MEK (mitogen-activated or extracellular signal-regulated protein kinase kinase) inhibitors. To identify potential mechanisms of acquired drug resistance, we generated clones resistant to the allosteric MEK inhibitor AZD6244 from two BRAF V600E mutant colorectal cancer cell lines that are highly sensitive to MEK or BRAF inhibition. These AZD6244-resistant (AR) clones, which exhibited cross-resistance to BRAF inhibitors, acquired resistance through amplification of the BRAF gene. A small percentage of treatment-naïve parental cells showed preexisting BRAF amplification. We observed similar amplification in a subset of cells in a BRAF-mutant colorectal cancer. In cell lines, BRAF amplification increased the abundance of phosphorylated MEK and impaired the ability of AZD6244 to inhibit ERK (extracellular signal-regulated kinase) phosphorylation. The ability of AZD6244 to inhibit ERK phosphorylation in AR cells was restored by treatment with a BRAF inhibitor at low concentrations that reduced the abundance of phosphorylated MEK to amounts observed in parental cells. Combined MEK and BRAF inhibition fully overcame resistance to MEK or BRAF inhibitors alone and was also more effective in parental cells compared to treatment with either inhibitor alone. These findings implicate BRAF amplification as a mechanism of resistance to both MEK and BRAF inhibitors and suggest combined MEK and BRAF inhibition as a clinical strategy to overcome, or possibly prevent, this mechanism of resistance.
Figures
References
-
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, Floyd Y, Gray K, Hall S, Hawes R, Hughes J, Kosmidou V, Menzies A, Mould C, Parker A, Stevens C, Watt S, Hooper S, Wilson R, Jayatilake H, Gusterson BA, Cooper C, Shipley J, Hargrave D, Pritchard-Jones K, Maitland N, Chenevix-Trench G, Riggins GJ, Bigner DD, Palmieri G, Cossu A, Flanagan A, Nicholson A, Ho JW, Leung SY, Yuen ST, Weber BL, Seigler HF, Darrow TL, Paterson H, Marais R, Marshall CJ, Wooster R, Stratton MR, Futreal PA. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. - PubMed
-
- McDermott U, Sharma SV, Dowell L, Greninger P, Montagut C, Lamb J, Archibald H, Raudales R, Tam A, Lee D, Rothenberg SM, Supko JG, Sordella R, Ulkus LE, Iafrate AJ, Maheswaran S, Njauw CN, Tsao H, Drew L, Hanke JH, Ma XJ, Erlander MG, Gray NS, Haber DA, Settleman J. Identification of genotype-correlated sensitivity to selective kinase inhibitors by using high-throughput tumor cell line profiling. Proc. Natl. Acad. Sci. U.S.A. 2007;104:19936–19941. - PMC - PubMed
-
- Rinehart J, Adjei AA, Lorusso PM, Waterhouse D, Hecht JR, Natale RB, Hamid O, Varterasian M, Asbury P, Kaldjian EP, Gulyas S, Mitchell DY, Herrera R, Sebolt-Leopold JS, Meyer MB. Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. J. Clin. Oncol. 2004;22:4456–4462. - PubMed
-
- Adjei AA, Cohen RB, Franklin W, Morris C, Wilson D, Molina JR, Hanson LJ, Gore L, Chow L, Leong S, Maloney L, Gordon G, Simmons H, Marlow A, Litwiler K, Brown S, Poch G, Kane K, Haney J, Eckhardt SG. Phase I pharmacokinetic and pharmacodynamic study of the oral, small-molecule mitogen-activated protein kinase kinase 1/2 inhibitor AZD6244 (ARRY-142886) in patients with advanced cancers. J. Clin. Oncol. 2008;26:2139–2146. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
