

Mre11-Rad50-Xrs2 and Sae2 promote 5' strand resection of DNA double-strand breaks
- PMID: 21102445
- PMCID: PMC3059534
- DOI: 10.1038/nsmb.1957
Mre11-Rad50-Xrs2 and Sae2 promote 5' strand resection of DNA double-strand breaks
Abstract
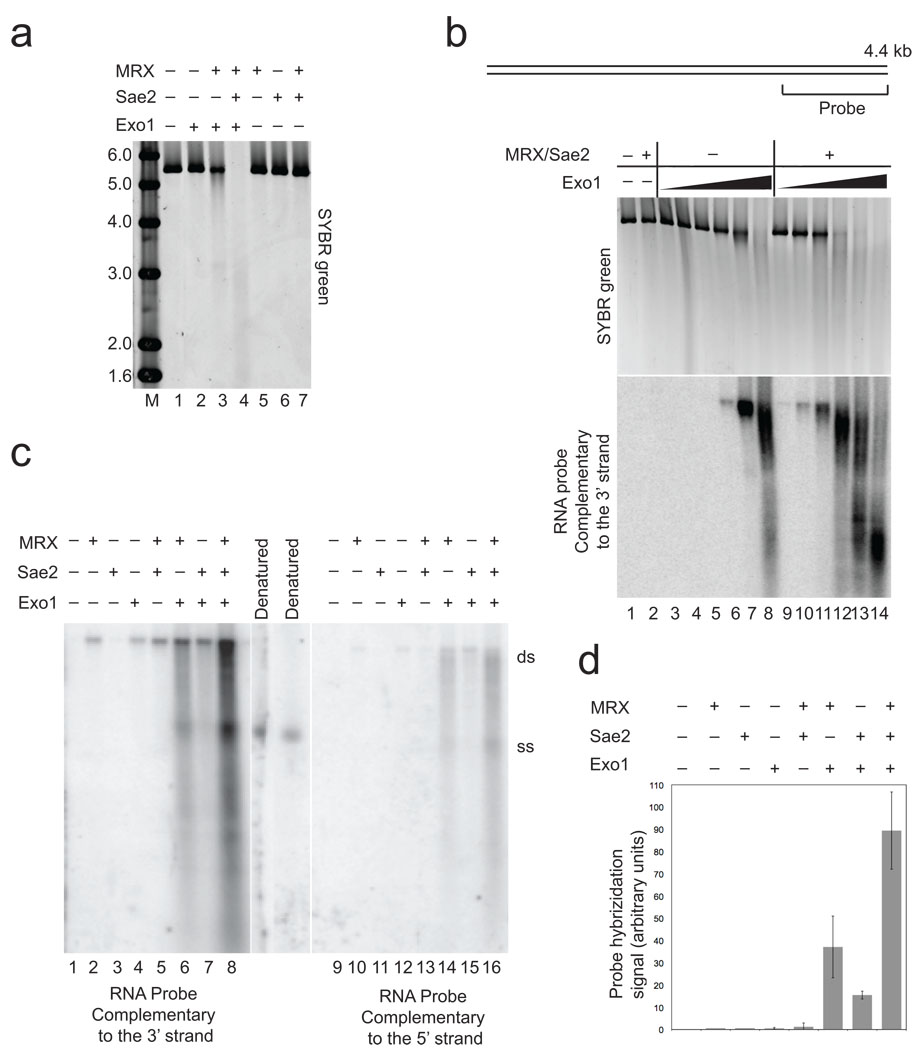
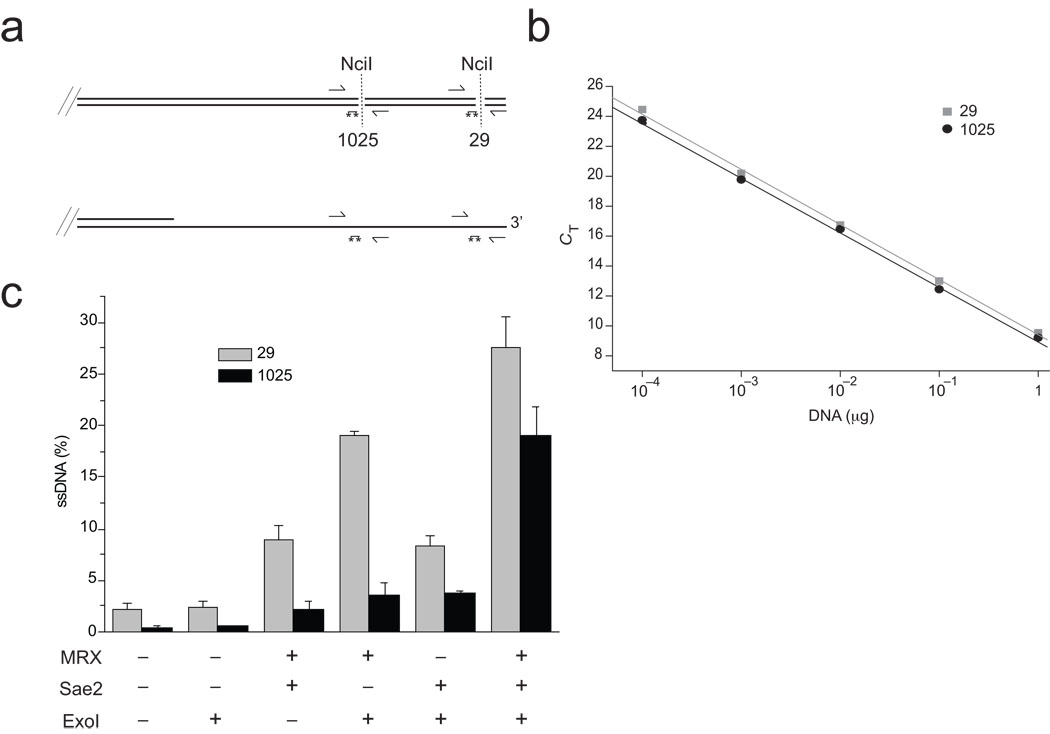
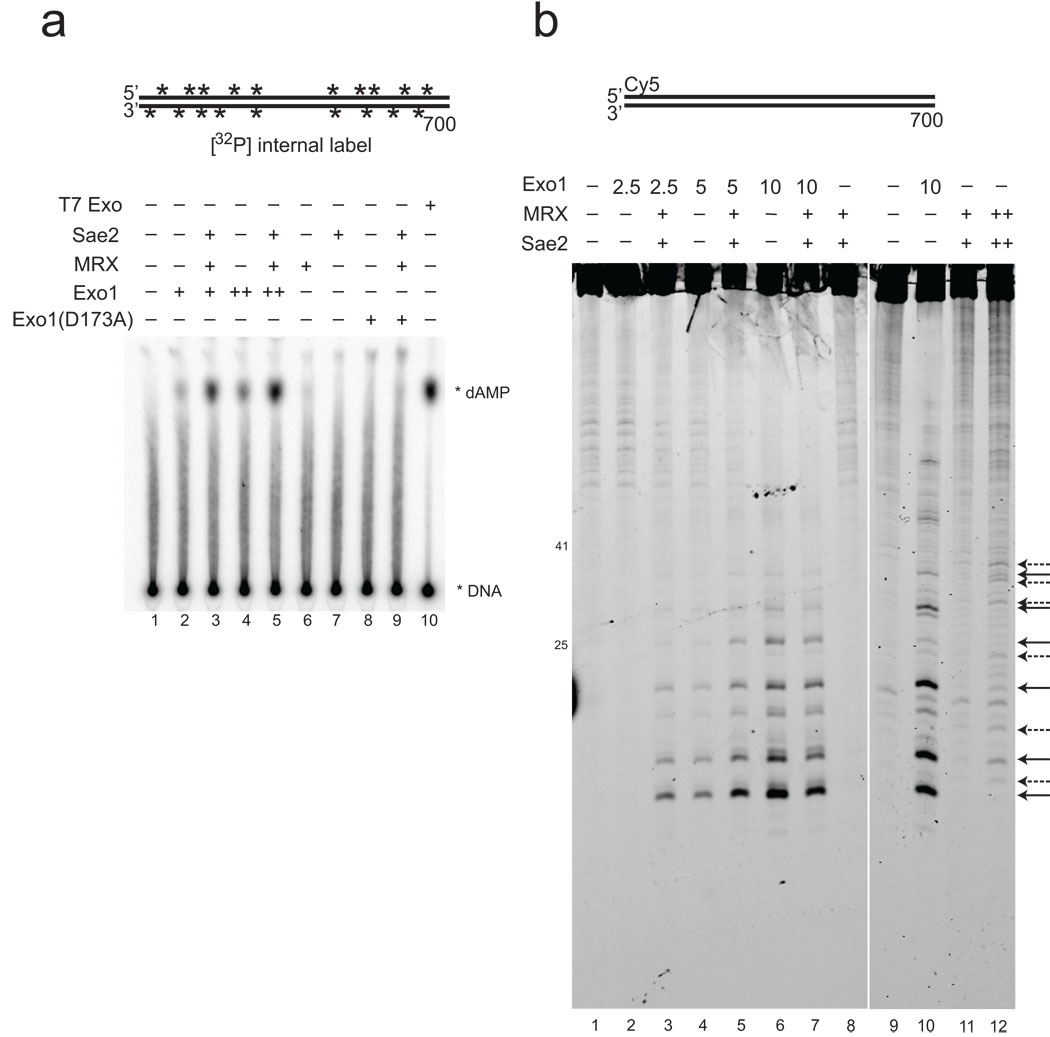
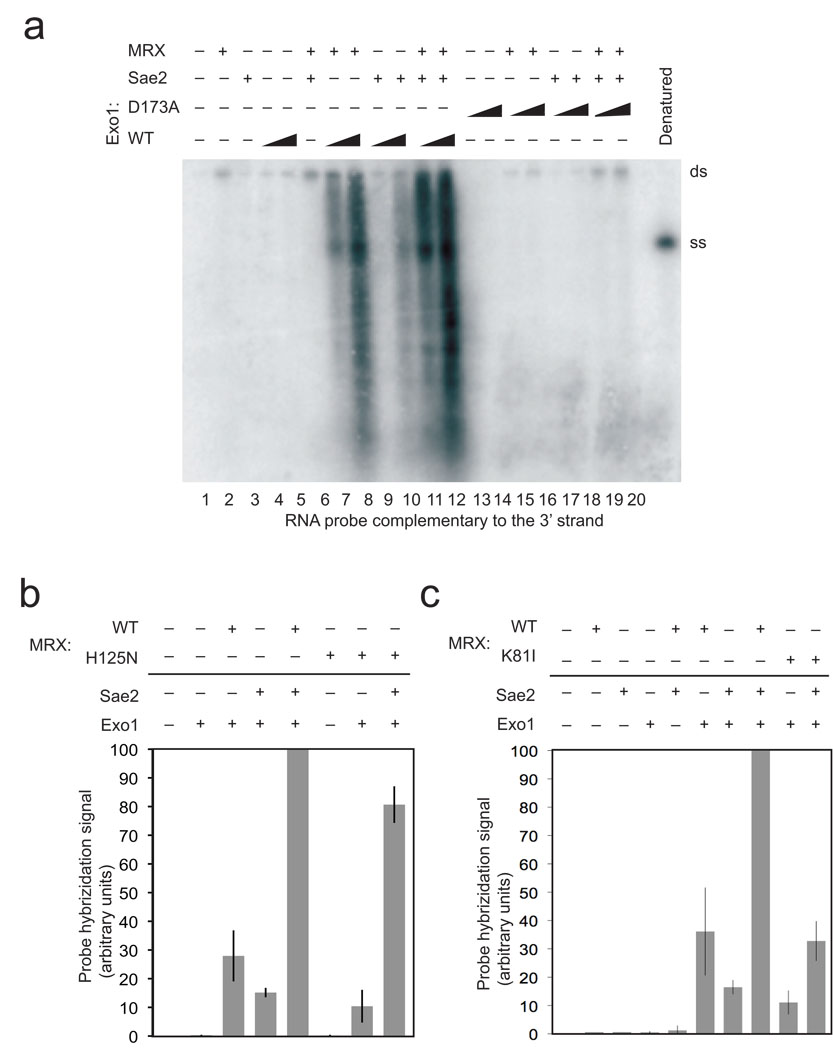
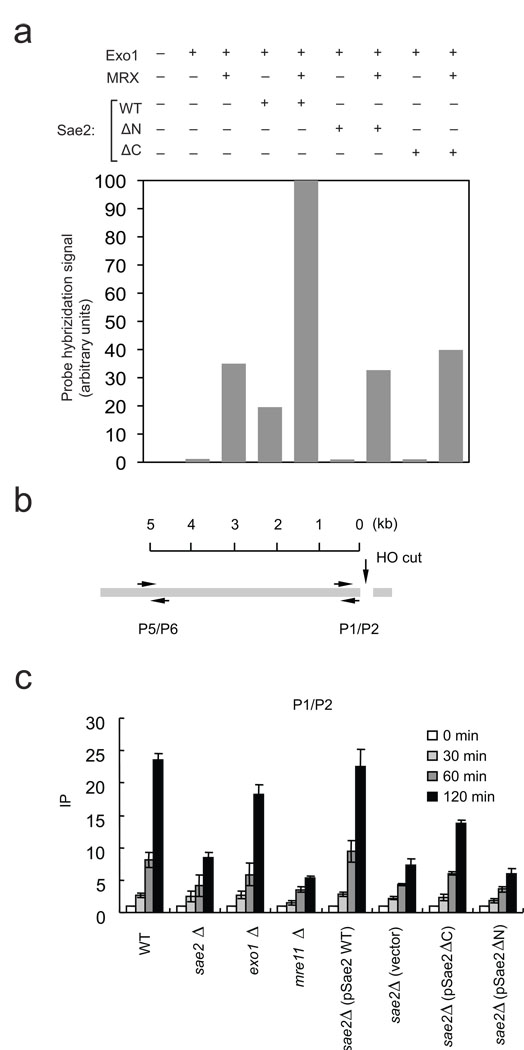
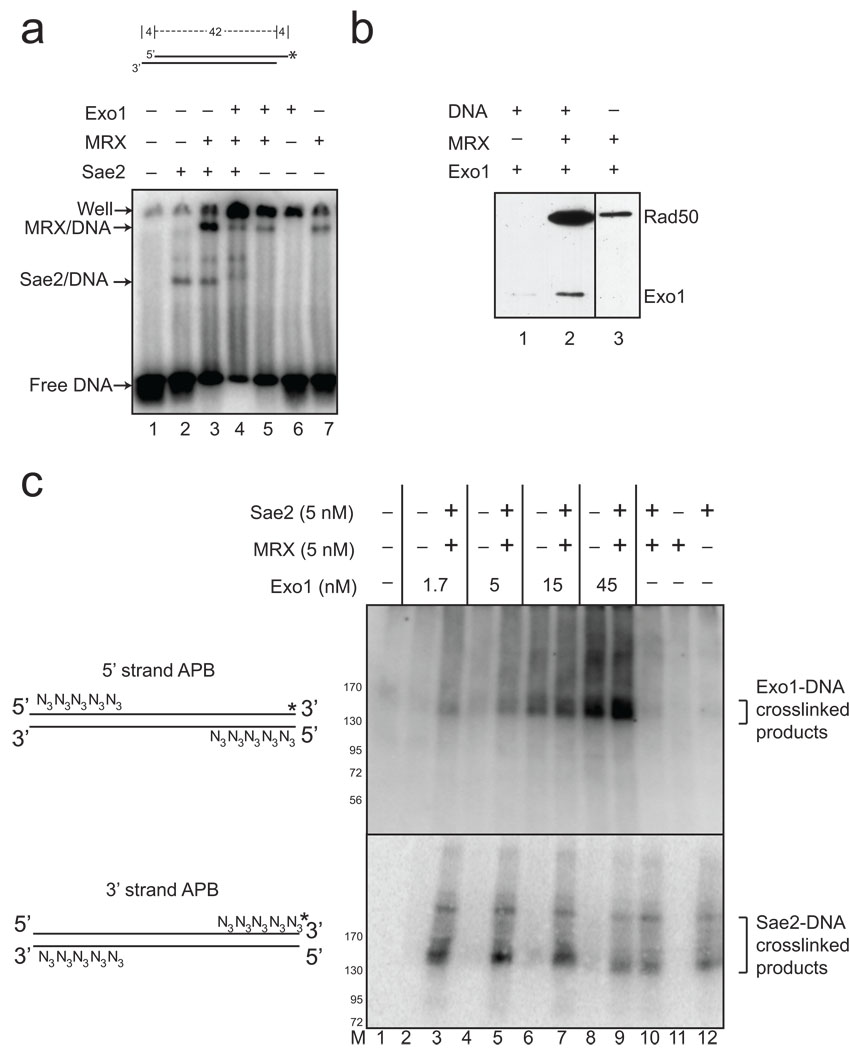
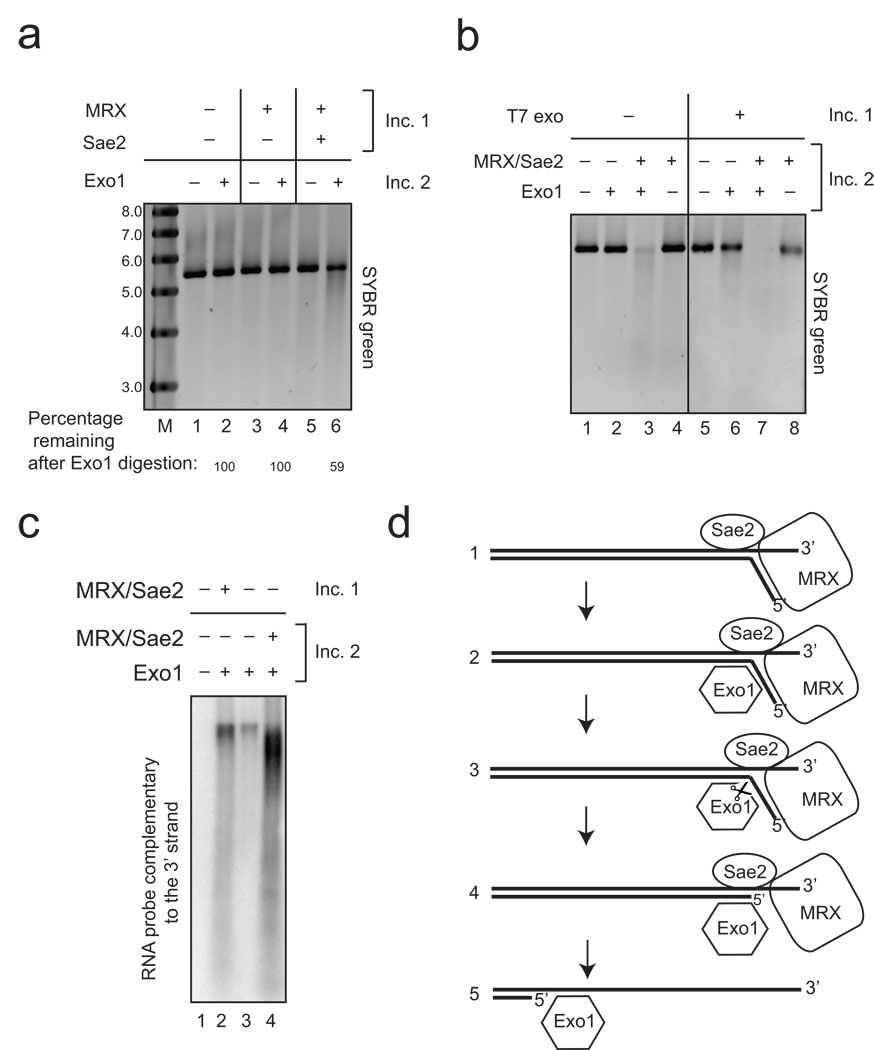
The repair of DNA double-strand breaks (DSBs) by homologous recombination is essential for genomic stability. The first step in this process is resection of 5' strands to generate 3' single-stranded DNA intermediates. Efficient resection in budding yeast requires the Mre11-Rad50-Xrs2 (MRX) complex and the Sae2 protein, although the role of MRX has been unclear because Mre11 paradoxically has 3'→5' exonuclease activity in vitro. Here we reconstitute resection with purified MRX, Sae2 and Exo1 proteins and show that degradation of the 5' strand is catalyzed by Exo1 yet completely dependent on MRX and Sae2 when Exo1 levels are limiting. This stimulation is mainly caused by cooperative binding of DNA substrates by Exo1, MRX and Sae2. This work establishes the direct role of MRX and Sae2 in promoting the resection of 5' strands in DNA DSB repair.
Figures
References
-
- San Filippo J, Sung P, Klein H. Mechanism of eukaryotic homologous recombination. Annu Rev Biochem. 2008;77:229–257. - PubMed
-
- Wyman C, Kanaar R. DNA double-strand break repair: all's well that ends well. Annu Rev Genet. 2006;40:363–383. - PubMed
-
- Krogh BO, Symington LS. Recombination proteins in yeast. Annu Rev Genet. 2004;38:233–271. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
