

Evaluation of external RNA controls for the standardisation of gene expression biomarker measurements
- PMID: 21106083
- PMCID: PMC3091780
- DOI: 10.1186/1471-2164-11-662
Evaluation of external RNA controls for the standardisation of gene expression biomarker measurements
Abstract
Background: Gene expression profiling is an important approach for detecting diagnostic and prognostic biomarkers, and predicting drug safety. The development of a wide range of technologies and platforms for measuring mRNA expression makes the evaluation and standardization of transcriptomic data problematic due to differences in protocols, data processing and analysis methods. Thus, universal RNA standards, such as those developed by the External RNA Controls Consortium (ERCC), are proposed to aid validation of research findings from diverse platforms such as microarrays and RT-qPCR, and play a role in quality control (QC) processes as transcriptomic profiling becomes more commonplace in the clinical setting.
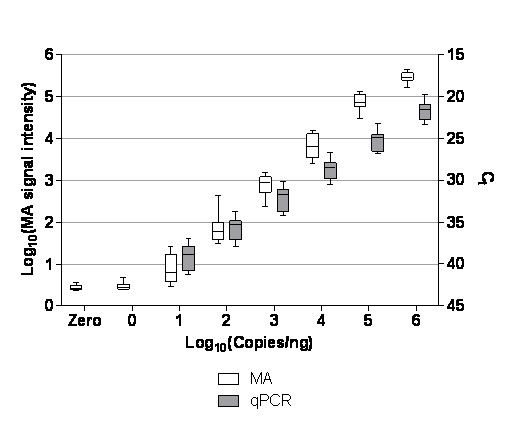
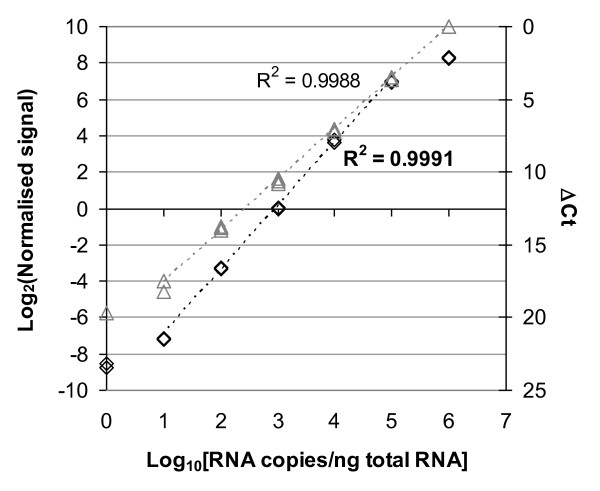
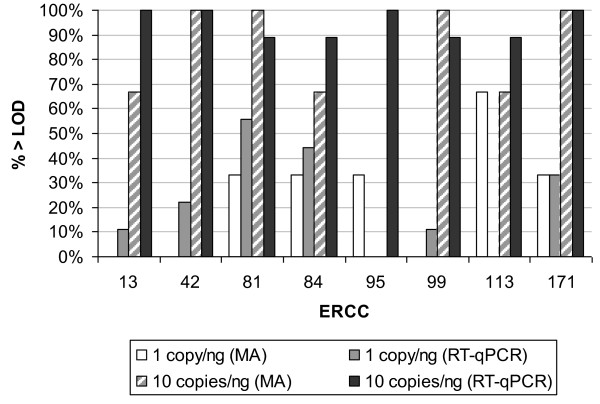
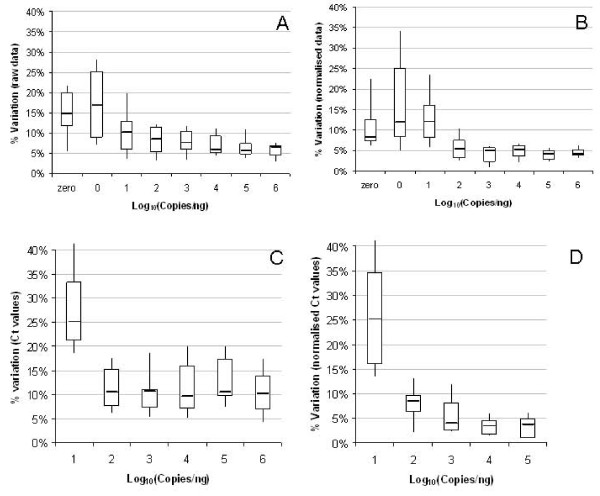
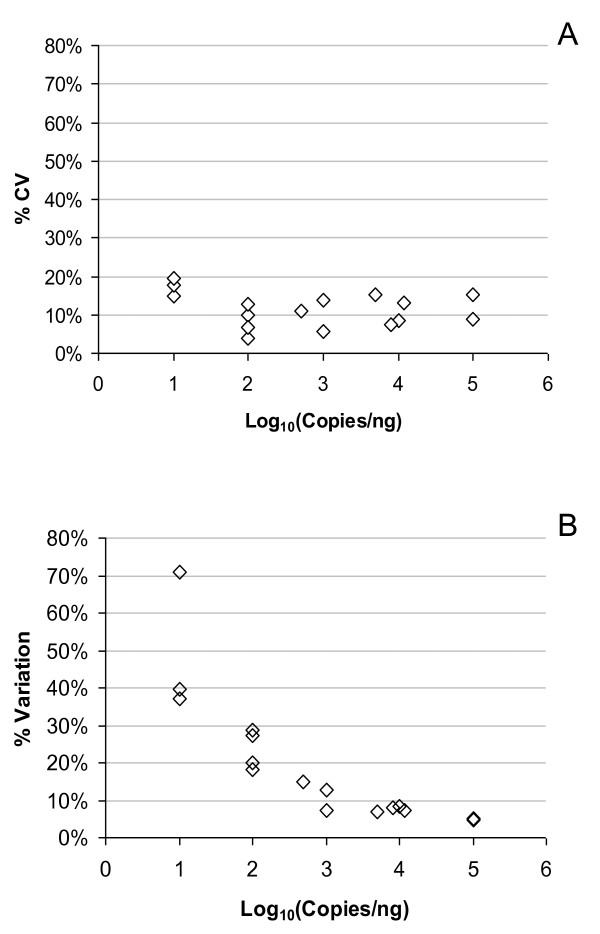
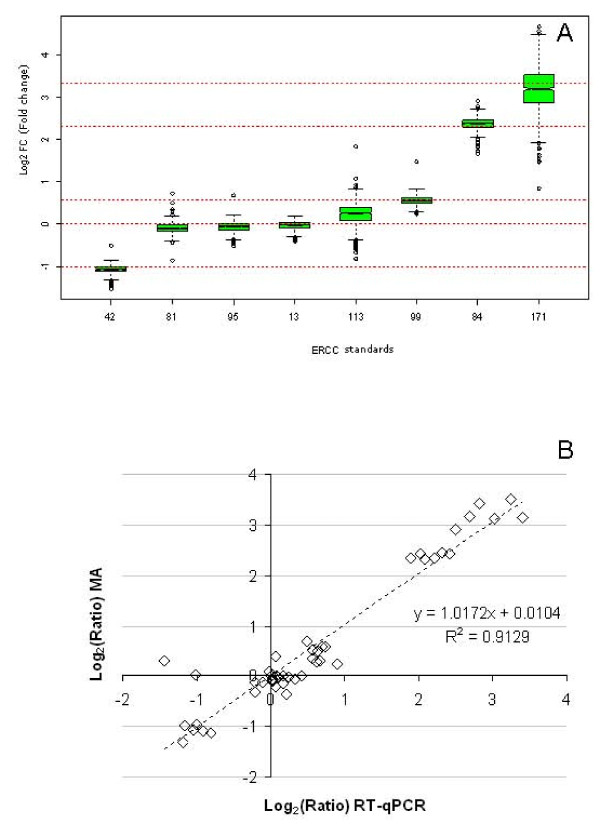
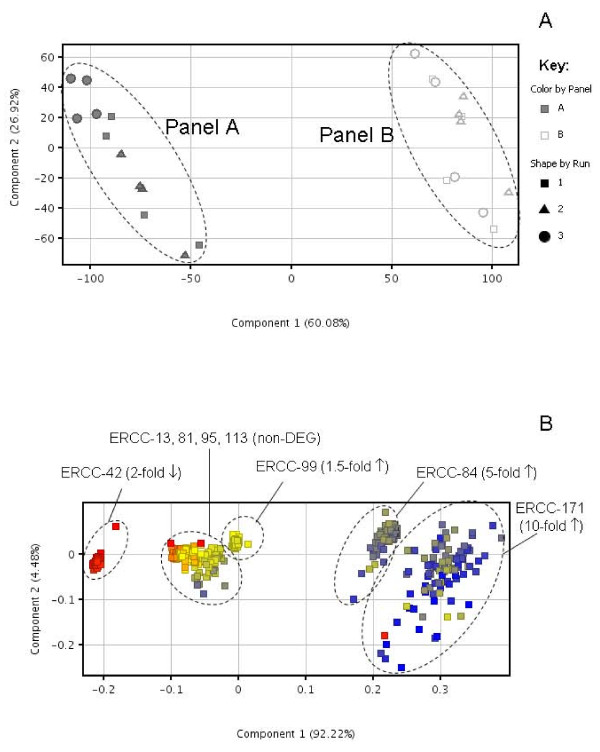
Results: Panels of ERCC RNA standards were constructed in order to test the utility of these reference materials (RMs) for performance characterization of two selected gene expression platforms, and for discrimination of biomarker profiles between groups. The linear range, limits of detection and reproducibility of microarray and RT-qPCR measurements were evaluated using panels of RNA standards. Transcripts of low abundance (≤ 10 copies/ng total RNA) showed more than double the technical variability compared to higher copy number transcripts on both platforms. Microarray profiling of two simulated 'normal' and 'disease' panels, each consisting of eight different RNA standards, yielded robust discrimination between the panels and between standards with varying fold change ratios, showing no systematic effects due to different labelling and hybridization runs. Also, comparison of microarray and RT-qPCR data for fold changes showed agreement for the two platforms.
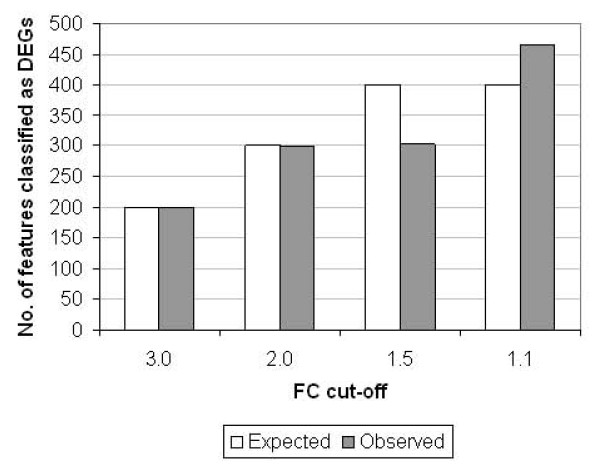
Conclusions: ERCC RNA standards provide a generic means of evaluating different aspects of platform performance, and can provide information on the technical variation associated with quantification of biomarkers expressed at different levels of physiological abundance. Distinct panels of standards serve as an ideal quality control tool kit for determining the accuracy of fold change cut-off threshold and the impact of experimentally-derived noise on the discrimination of normal and disease profiles.
Figures
References
-
- Waring JF, Halbert DN. The promise of toxicogenomics. Curr Opin Mol Ther. 2002;4:229–235. - PubMed
-
- Buyse M, Loi S, van't Veer L, Viale G, Delorenzi M, Glas AM, Saghatchian d'Assignies M, Bergh J, Lidereau R, Ellis P. et al. Validation and Clinical Utility of a 70-Gene Prognostic Signature for Women With Node-Negative Breast Cancer. J Natl Cancer Inst. 2006;98:1183–1192. doi: 10.1093/jnci/djj329. - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
