

New horizons in the treatment of cystic fibrosis
- PMID: 21108631
- PMCID: PMC3085876
- DOI: 10.1111/j.1476-5381.2010.01137.x
New horizons in the treatment of cystic fibrosis
Abstract
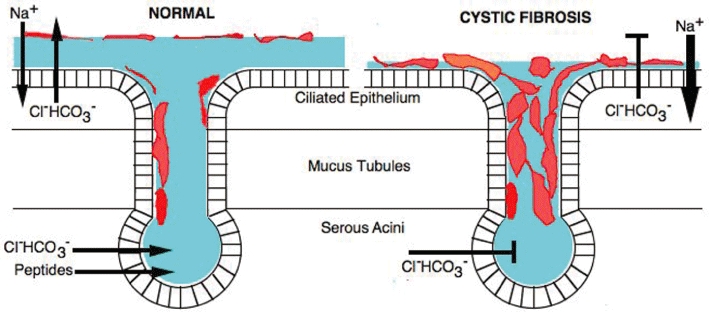
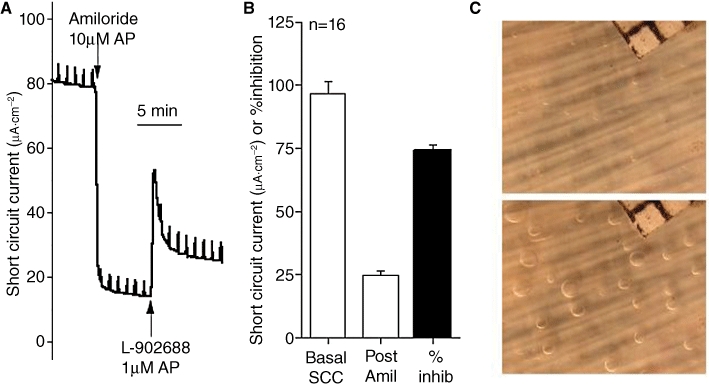
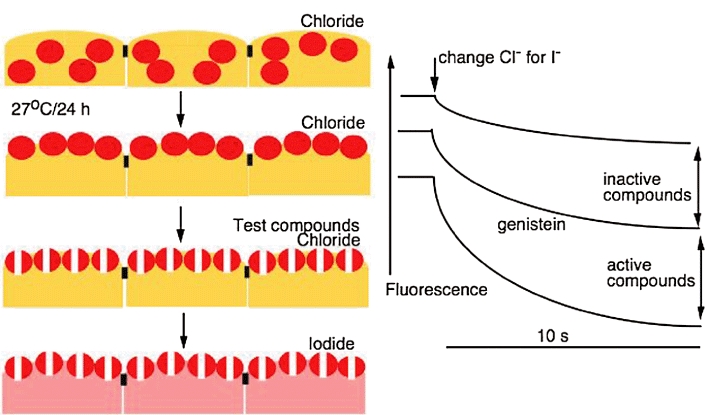
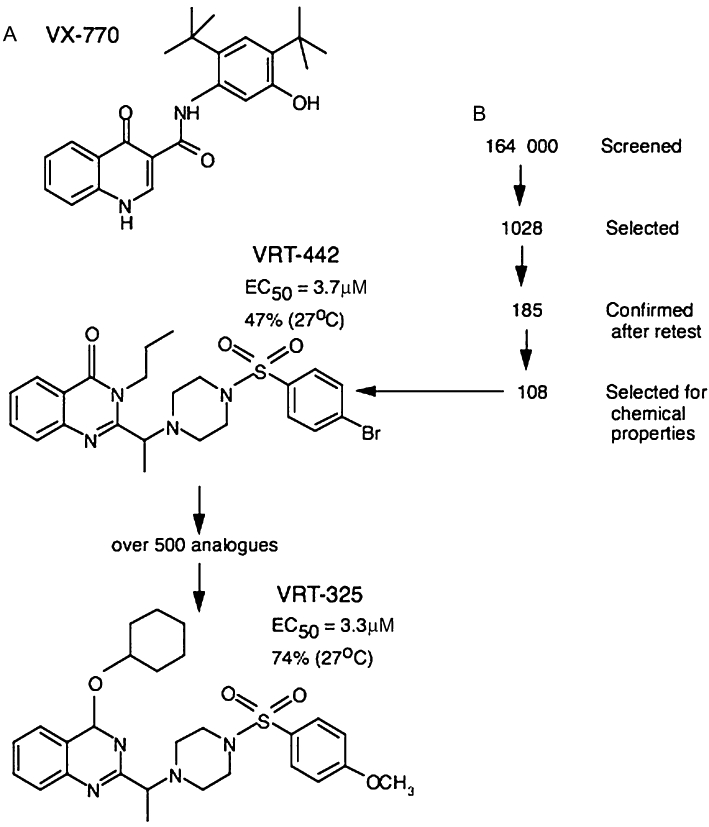
Cystic fibrosis (CF) is a lethal, recessive, genetic disease affecting approximately 1 in 2500 live births among Caucasians. The CF gene codes for a cAMP/PKA-dependent, ATP-requiring, membrane chloride ion channel, generally found in the apical membranes of many secreting epithelia and known as CFTR (cystic fibrosis transmembrane conductance regulator). There are currently over 1700 known mutations affecting CFTR, many of which give rise to a disease phenotype. Around 75% of CF alleles contain the ΔF508 mutation in which a triplet codon has been lost, leading to a missing phenylalanine at position 508 in the protein. This altered protein fails to be trafficked to the correct location in the cell and is generally destroyed by the proteasome. The small amount that does reach the correct location functions poorly. Clearly the cohort of patients with at least one ΔF508 allele are a major target for therapeutic intervention. It is now over two decades since the CF gene was discovered and during this time the properties of CFTR have been intensely investigated. At long last there appears to be progress with the pharmaco-therapeutic approach. Ongoing clinical trials have produced fascinating results in which clinical benefit appears to have been achieved. To arrive at this point ingenious ways have been devised to screen very large chemical libraries for one of two properties: (i) agents promoting trafficking of mutant CFTR to, and insertion into the membrane, and known as correctors or (ii) agents which activate appropriately located mutant CFTR, known as potentiators. The best compounds emerging from these programmes are then used as chemical scaffolds to synthesize other compounds with appropriate pharmaceutical properties, hopefully with their pharmacological activity maintained or even enhanced. In summary, this approach attempts to make the mutant CFTR function in place of the real CFTR. A major function of CFTR in healthy airways is to maintain an adequate airway surface liquid (ASL) layer. In CF the position is further confounded since epithelial sodium channels (ENaC) are no longer regulated and transport salt and water out of the airways to exacerbate the lack of ASL. Thus an additional possibility for treatment of CF is to use agents that inhibit ENaC either alone or as adjuncts to CFTR correctors and/or potentiators. Yet a further way in which a pharmacological approach to CF can be considered is to recruit alternative chloride channels, such as calcium-activated chloride channel (CaCC), to act as surrogates for CFTR. A number of P2Y(2) receptor agonists have been investigated that operate by increasing Ca(2+)(i) which in turn activates CaCC. Some of these compounds are currently in clinical trials. The knowledge base surrounding the structure and function of CFTR that has accumulated in the last 20 years is impressive. Translational research feeding from this is now yielding compounds that provide real prospects for a pharmacotherapy for this disease.
© 2010 The Author. British Journal of Pharmacology © 2010 The British Pharmacological Society.
Figures
Similar articles
-
Pharmacotherapy of the ion transport defect in cystic fibrosis: role of purinergic receptor agonists and other potential therapeutics.Am J Respir Med. 2003;2(4):299-309. doi: 10.1007/BF03256658. Am J Respir Med. 2003. PMID: 14719996 Review.
-
Low temperature and chemical rescue affect molecular proximity of DeltaF508-cystic fibrosis transmembrane conductance regulator (CFTR) and epithelial sodium channel (ENaC).J Biol Chem. 2012 May 11;287(20):16781-90. doi: 10.1074/jbc.M111.332031. Epub 2012 Mar 22. J Biol Chem. 2012. PMID: 22442149 Free PMC article.
-
Pharmacological treatment of the ion transport defect in cystic fibrosis.Expert Opin Investig Drugs. 2001 Jan;10(1):1-19. doi: 10.1517/13543784.10.1.1. Expert Opin Investig Drugs. 2001. PMID: 11116277 Review.
-
Targeting autophagy as a novel strategy for facilitating the therapeutic action of potentiators on ΔF508 cystic fibrosis transmembrane conductance regulator.Autophagy. 2012 Nov;8(11):1657-72. doi: 10.4161/auto.21483. Epub 2012 Aug 9. Autophagy. 2012. PMID: 22874563 Free PMC article.
-
Small-molecule drugs for cystic fibrosis: Where are we now?Pulm Pharmacol Ther. 2022 Feb;72:102098. doi: 10.1016/j.pupt.2021.102098. Epub 2021 Nov 15. Pulm Pharmacol Ther. 2022. PMID: 34793977 Review.
Cited by
-
Pulmonary Function and Hospital Admission in Patients with Cystic Fibrosis Based on Household Second-Hand Smoking.Tanaffos. 2018 Jan;17(1):37-41. Tanaffos. 2018. PMID: 30116277 Free PMC article.
-
Calcium-calmodulin does not alter the anion permeability of the mouse TMEM16A calcium-activated chloride channel.J Gen Physiol. 2014 Jul;144(1):115-24. doi: 10.1085/jgp.201411179. J Gen Physiol. 2014. PMID: 24981232 Free PMC article.
-
Esc peptides as novel potentiators of defective cystic fibrosis transmembrane conductance regulator: an unprecedented property of antimicrobial peptides.Cell Mol Life Sci. 2021 Dec 31;79(1):67. doi: 10.1007/s00018-021-04030-2. Cell Mol Life Sci. 2021. PMID: 34971429 Free PMC article.
-
Biochemical and biophysical characterization of inositol-tetrakisphosphate 1-kinase inhibitors.J Biol Chem. 2025 Mar;301(3):108274. doi: 10.1016/j.jbc.2025.108274. Epub 2025 Feb 6. J Biol Chem. 2025. PMID: 39922495 Free PMC article.
-
The road for survival improvement of cystic fibrosis patients in Arab countries.Int J Pediatr Adolesc Med. 2015 Jun;2(2):47-58. doi: 10.1016/j.ijpam.2015.05.006. Epub 2015 Jun 19. Int J Pediatr Adolesc Med. 2015. PMID: 30805437 Free PMC article. Review.
References
-
- Accurso FJ, Rowe SM, Durie PR, Konstan MW, Dunitz J, Hornick DB, et al. Interim results of a phase 2a study of VX-770 to evaluate safety, pharmacokinetics and biomarkers of CFTR activity in cystic fibrosis subjects with G551D. Pediatr Pulmonol Suppl. 2008;31:295.
-
- Adam G, Ousingsawat J, Schreiber R, Kunzelmann K. Increase in intracellular Cl- concentration by cAMP and Ca2+-dependent stimulation of M1 collecting duct cells. Eur J Physiol. 2005;449:470–478. - PubMed
-
- Ballard ST, Taylor AE. Bioelectric properties of proximal bronchiolar epithelium. Am J Physiol. 1994;267:79–84. - PubMed
-
- Ballard ST, Trout L, Bebok Z, Sorscher EJ, Crews A. CFTR involvement in chloride, bicarbonate, and liquid secretion by airway submucosal glands. Am J Physiol. 1999;277:L694–L699. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
