

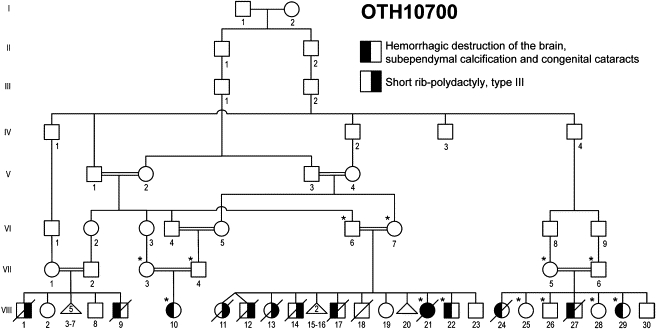
A homozygous mutation in the tight-junction protein JAM3 causes hemorrhagic destruction of the brain, subependymal calcification, and congenital cataracts
- PMID: 21109224
- PMCID: PMC2997371
- DOI: 10.1016/j.ajhg.2010.10.026
A homozygous mutation in the tight-junction protein JAM3 causes hemorrhagic destruction of the brain, subependymal calcification, and congenital cataracts
Abstract
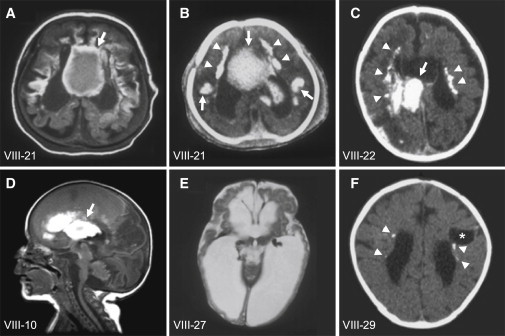
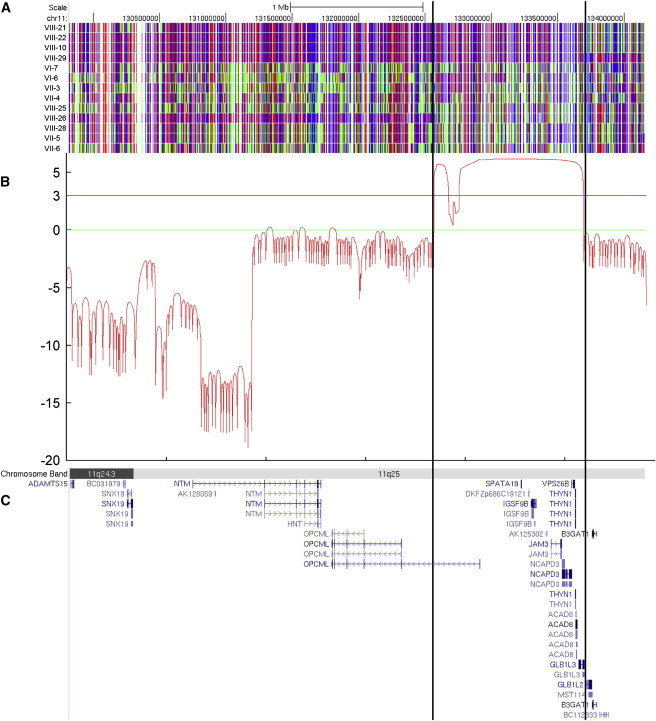
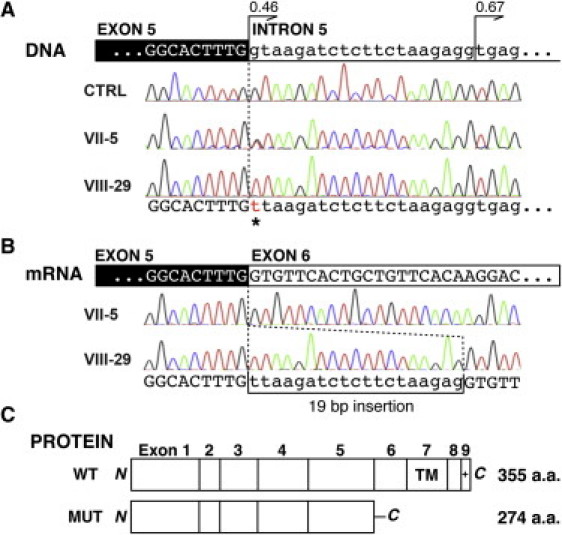
The tight junction, or zonula occludens, is a specialized cell-cell junction that regulates epithelial and endothelial permeability, and it is an essential component of the blood-brain barrier in the cerebrovascular endothelium. In addition to functioning as a diffusion barrier, tight junctions are also involved in signal transduction. In this study, we identified a homozygous mutation in the tight-junction protein gene JAM3 in a large consanguineous family from the United Arab Emirates. Some members of this family had a rare autosomal-recessive syndrome characterized by severe hemorrhagic destruction of the brain, subependymal calcification, and congenital cataracts. Their clinical presentation overlaps with some reported cases of pseudo-TORCH syndrome as well as with cases involving mutations in occludin, another component of the tight-junction complex. However, massive intracranial hemorrhage distinguishes these patients from others. Homozygosity mapping identified the disease locus in this family on chromosome 11q25 with a maximum multipoint LOD score of 6.15. Sequence analysis of genes in the candidate interval uncovered a mutation in the canonical splice-donor site of intron 5 of JAM3. RT-PCR analysis of a patient lymphoblast cell line confirmed abnormal splicing, leading to a frameshift mutation with early termination. JAM3 is known to be present in vascular endothelium, although its roles in cerebral vasculature have not been implicated. Our results suggest that JAM3 is essential for maintaining the integrity of the cerebrovascular endothelium as well as for normal lens development in humans.
Copyright © 2010 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Vestweber D. Molecular mechanisms that control endothelial cell contacts. J. Pathol. 2000;190:281–291. - PubMed
-
- Carlton V.E., Harris B.Z., Puffenberger E.G., Batta A.K., Knisely A.S., Robinson D.L., Strauss K.A., Shneider B.L., Lim W.A., Salen G. Complex inheritance of familial hypercholanemia with associated mutations in TJP2 and BAAT. Nat. Genet. 2003;34:91–96. - PubMed
-
- Wilcox E.R., Burton Q.L., Naz S., Riazuddin S., Smith T.N., Ploplis B., Belyantseva I., Ben-Yosef T., Liburd N.A., Morell R.J. Mutations in the gene encoding tight junction claudin-14 cause autosomal recessive deafness DFNB29. Cell. 2001;104:165–172. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
