

Whole-exome-sequencing-based discovery of human FADD deficiency
- PMID: 21109225
- PMCID: PMC2997374
- DOI: 10.1016/j.ajhg.2010.10.028
Whole-exome-sequencing-based discovery of human FADD deficiency
Abstract
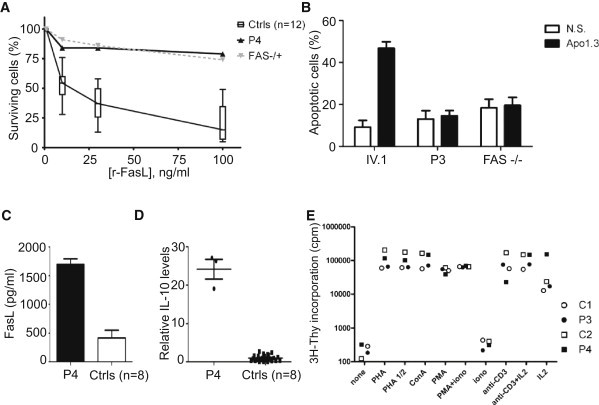
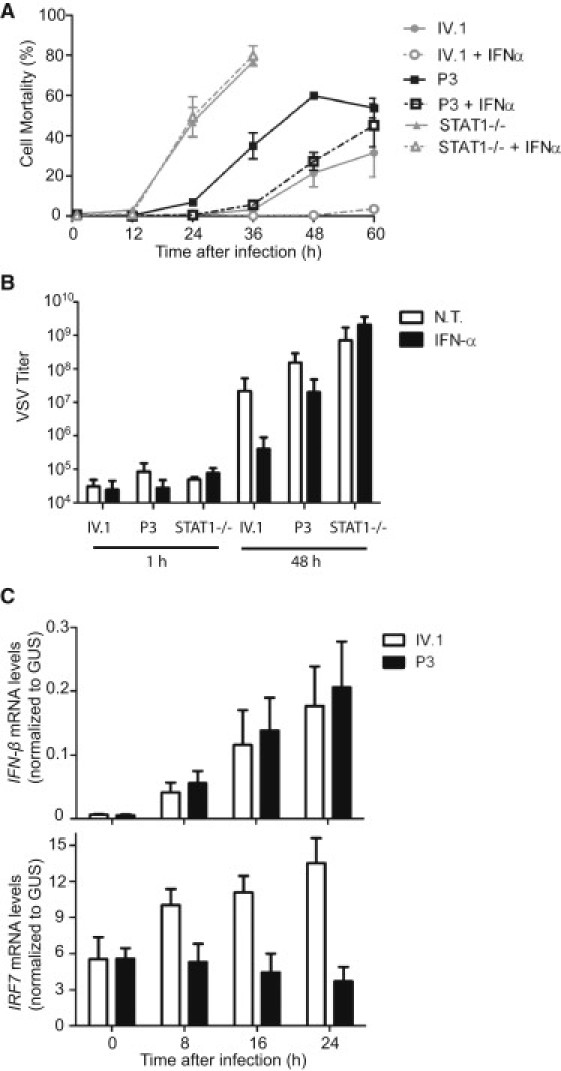
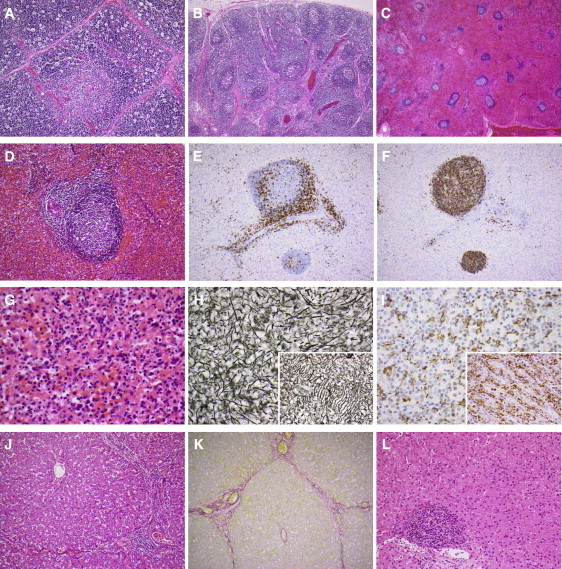
Germline mutations in FASL and FAS impair Fas-dependent apoptosis and cause recessively or dominantly inherited autoimmune lymphoproliferative syndrome (ALPS). Patients with ALPS typically present with no other clinical phenotype. We investigated a large, consanguineous, multiplex kindred in which biological features of ALPS were found in the context of severe bacterial and viral disease, recurrent hepatopathy and encephalopathy, and cardiac malformations. By a combination of genome-wide linkage and whole-exome sequencing, we identified a homozygous missense mutation in FADD, encoding the Fas-associated death domain protein (FADD), in the patients. This FADD mutation decreases steady-state protein levels and impairs Fas-dependent apoptosis in vitro, accounting for biological ALPS phenotypes in vivo. It also impairs Fas-independent signaling pathways. The observed bacterial infections result partly from functional hyposplenism, and viral infections result from impaired interferon immunity. We describe here a complex clinical disorder, its genetic basis, and some of the key mechanisms underlying its pathogenesis. Our findings highlight the key role of FADD in Fas-dependent and Fas-independent signaling pathways in humans.
Copyright © 2010 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures

References
-
- Del-Rey M., Ruiz-Contreras J., Bosque A., Calleja S., Gomez-Rial J., Roldan E., Morales P., Serrano A., Anel A., Paz-Artal E., Allende L.M. A homozygous Fas ligand gene mutation in a patient causes a new type of autoimmune lymphoproliferative syndrome. Blood. 2006;108:1306–1312. - PubMed
-
- Fisher G.H., Rosenberg F.J., Straus S.E., Dale J.K., Middleton L.A., Lin A.Y., Strober W., Lenardo M.J., Puck J.M. Dominant interfering Fas gene mutations impair apoptosis in a human autoimmune lymphoproliferative syndrome. Cell. 1995;81:935–946. - PubMed
-
- Rieux-Laucat F., Le Deist F., Hivroz C., Roberts I.A., Debatin K.M., Fischer A., de Villartay J.P. Mutations in Fas associated with human lymphoproliferative syndrome and autoimmunity. Science. 1995;268:1347–1349. - PubMed
-
- Casanova J.L., Abel L. Primary immunodeficiencies: a field in its infancy. Science. 2007;317:617–619. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
