

A human genome structural variation sequencing resource reveals insights into mutational mechanisms
- PMID: 21111241
- PMCID: PMC3026629
- DOI: 10.1016/j.cell.2010.10.027
A human genome structural variation sequencing resource reveals insights into mutational mechanisms
Abstract
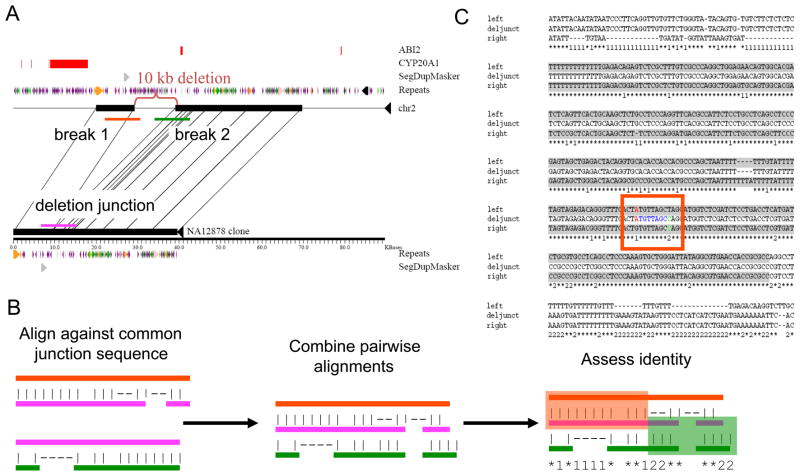
Understanding the prevailing mutational mechanisms responsible for human genome structural variation requires uniformity in the discovery of allelic variants and precision in terms of breakpoint delineation. We develop a resource based on capillary end sequencing of 13.8 million fosmid clones from 17 human genomes and characterize the complete sequence of 1054 large structural variants corresponding to 589 deletions, 384 insertions, and 81 inversions. We analyze the 2081 breakpoint junctions and infer potential mechanism of origin. Three mechanisms account for the bulk of germline structural variation: microhomology-mediated processes involving short (2-20 bp) stretches of sequence (28%), nonallelic homologous recombination (22%), and L1 retrotransposition (19%). The high quality and long-range continuity of the sequence reveals more complex mutational mechanisms, including repeat-mediated inversions and gene conversion, that are most often missed by other methods, such as comparative genomic hybridization, single nucleotide polymorphism microarrays, and next-generation sequencing.
Copyright © 2010 Elsevier Inc. All rights reserved.
Conflict of interest statement
E.E.E is on the scientific advisory board for Pacific Biosciences. T.L.N. is an employee and founder of iGenix Inc.
Figures





References
-
- Abe H, Ochi H, Maekawa T, Hatakeyama T, Tsuge M, Kitamura S, Kimura T, Miki D, Mitsui F, Hiraga N, et al. Effects of structural variations of APOBEC3A and APOBEC3B genes in chronic hepatitis B virus infection. Hepatol Res. 2009;39:1159–1168. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
