

Mitochondrial optic neuropathies - disease mechanisms and therapeutic strategies
- PMID: 21112411
- PMCID: PMC3081075
- DOI: 10.1016/j.preteyeres.2010.11.002
Mitochondrial optic neuropathies - disease mechanisms and therapeutic strategies
Abstract
Leber hereditary optic neuropathy (LHON) and autosomal-dominant optic atrophy (DOA) are the two most common inherited optic neuropathies in the general population. Both disorders share striking pathological similarities, marked by the selective loss of retinal ganglion cells (RGCs) and the early involvement of the papillomacular bundle. Three mitochondrial DNA (mtDNA) point mutations; m.3460G>A, m.11778G>A, and m.14484T>C account for over 90% of LHON cases, and in DOA, the majority of affected families harbour mutations in the OPA1 gene, which codes for a mitochondrial inner membrane protein. Optic nerve degeneration in LHON and DOA is therefore due to disturbed mitochondrial function and a predominantly complex I respiratory chain defect has been identified using both in vitro and in vivo biochemical assays. However, the trigger for RGC loss is much more complex than a simple bioenergetic crisis and other important disease mechanisms have emerged relating to mitochondrial network dynamics, mtDNA maintenance, axonal transport, and the involvement of the cytoskeleton in maintaining a differential mitochondrial gradient at sites such as the lamina cribosa. The downstream consequences of these mitochondrial disturbances are likely to be influenced by the local cellular milieu. The vulnerability of RGCs in LHON and DOA could derive not only from tissue-specific, genetically-determined biological factors, but also from an increased susceptibility to exogenous influences such as light exposure, smoking, and pharmacological agents with putative mitochondrial toxic effects. Our concept of inherited mitochondrial optic neuropathies has evolved over the past decade, with the observation that patients with LHON and DOA can manifest a much broader phenotypic spectrum than pure optic nerve involvement. Interestingly, these phenotypes are sometimes clinically indistinguishable from other neurodegenerative disorders such as Charcot-Marie-Tooth disease, hereditary spastic paraplegia, and multiple sclerosis, where mitochondrial dysfunction is also thought to be an important pathophysiological player. A number of vertebrate and invertebrate disease models has recently been established to circumvent the lack of human tissues, and these have already provided considerable insight by allowing direct RGC experimentation. The ultimate goal is to translate these research advances into clinical practice and new treatment strategies are currently being investigated to improve the visual prognosis for patients with mitochondrial optic neuropathies.
Copyright © 2010 Elsevier Ltd. All rights reserved.
Figures
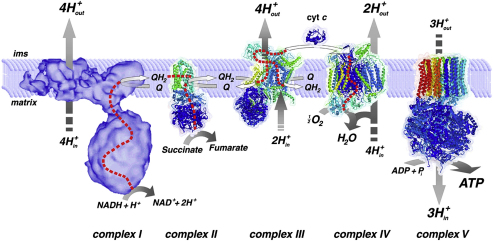
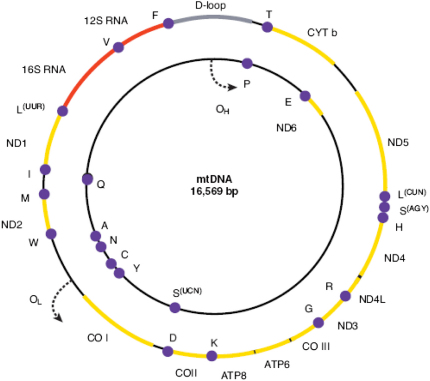




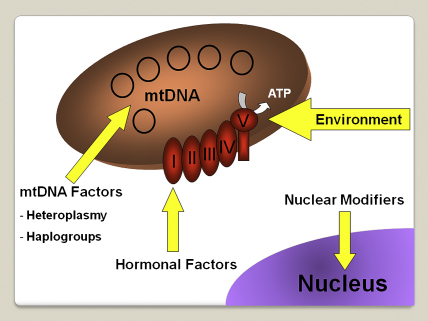
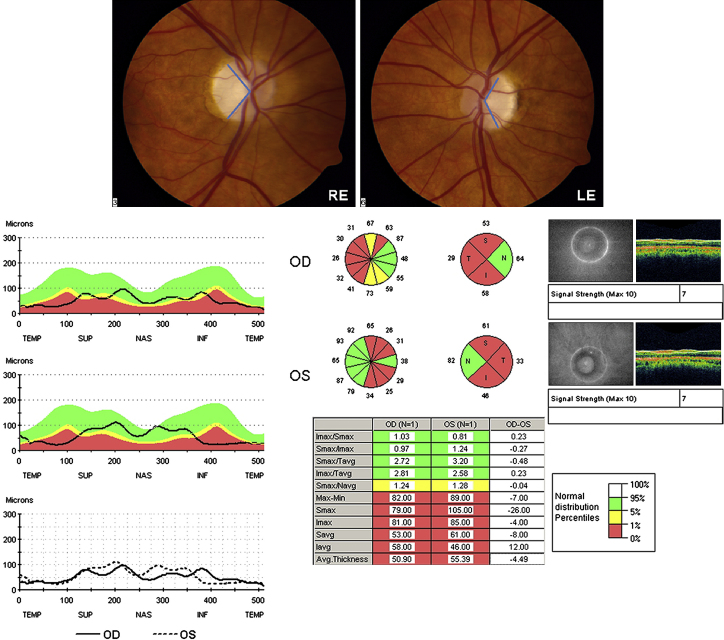
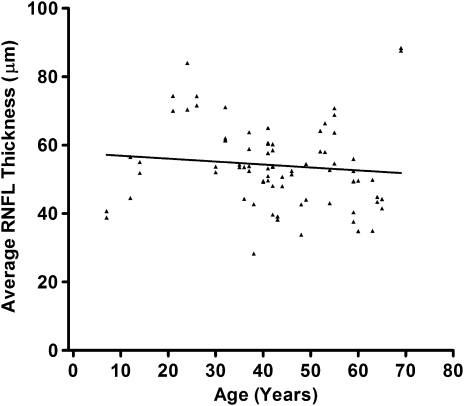
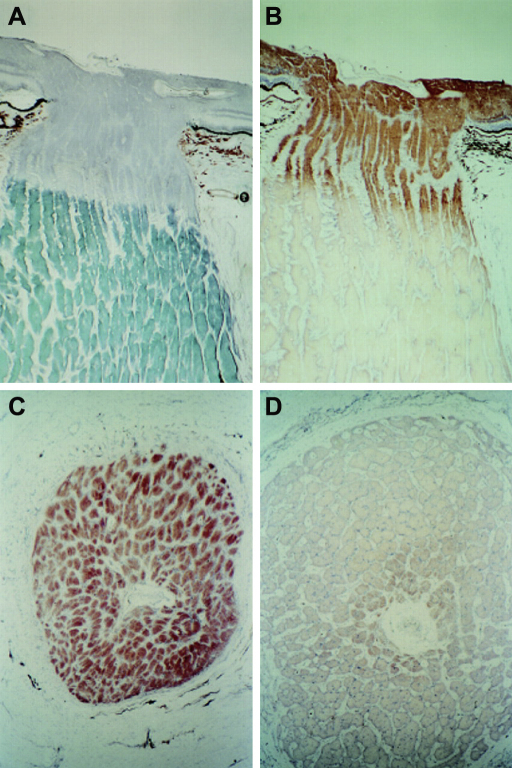
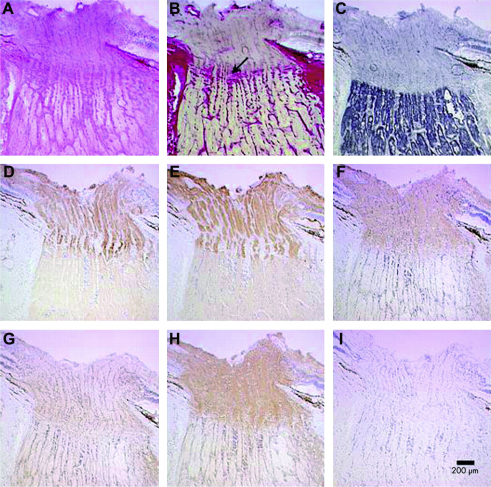

Comment in
-
Parsing the differences in affected with LHON: genetic versus environmental triggers of disease conversion.Brain. 2016 Mar;139(Pt 3):e17. doi: 10.1093/brain/awv339. Epub 2015 Dec 10. Brain. 2016. PMID: 26657166 Free PMC article. No abstract available.
-
Reply: Parsing the differences in affected with LHON: genetic versus environmental triggers of disease conversion.Brain. 2016 Mar;139(Pt 3):e18. doi: 10.1093/brain/awv340. Epub 2015 Dec 10. Brain. 2016. PMID: 26657167 Free PMC article. No abstract available.
References
-
- Ahn J., Kim N.J., Choung H.K., Hwang S.W., Sung M., Lee M.J. Frontalis sling operation using silicone rod for the correction of ptosis in chronic progressive external ophthalmoplegia. British Journal of Ophthalmology. 2008;92:1685–1688. - PubMed
-
- Ahrlich K.G., De Moraes C.G.V., Teng C.C., Prata T.S., Tello C., Ritch R. Visual field progression differences between normal-tension and exfoliative high-tension glaucoma. Investigative Ophthalmology & Visual Science. 2010;51:1458–1463. - PubMed
-
- Aijaz S., Erskine L., Jeffery G., Bhattacharya S.S., Votruba M. Developmental expression profile of the optic atrophy gene product: OPA1 is not localized exclusively in the mammalian retinal ganglion cell layer. Investigative Ophthalmology & Visual Science. 2004;45:1667–1673. - PubMed
-
- Akepati V.R. Characterization of OPA1 isoforms isolated from mouse tissues. Journal of Neurochemistry. 2008;106:372–383. - PubMed
-
- Alavi M.V., Bette S., Schimpf S., Schuettauf F., Schraermeyer U., Wehrl H.F. A splice site mutation in the murine Opa1 gene features pathology of autosomal dominant optic atrophy. Brain. 2007;130:1029–1042. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
