

Cystic diseases of the kidney: ciliary dysfunction and cystogenic mechanisms
- PMID: 21113628
- PMCID: PMC3640323
- DOI: 10.1007/s00467-010-1697-5
Cystic diseases of the kidney: ciliary dysfunction and cystogenic mechanisms
Abstract
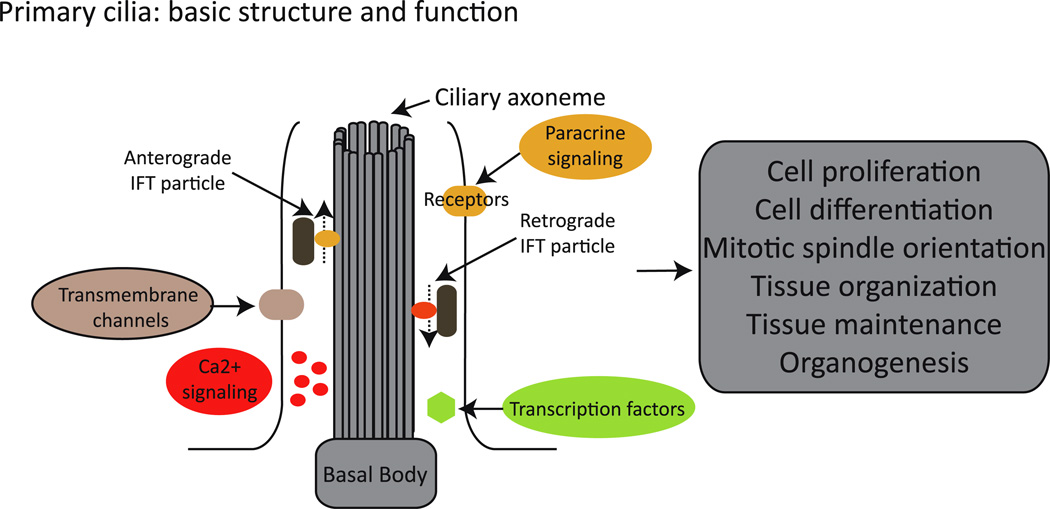
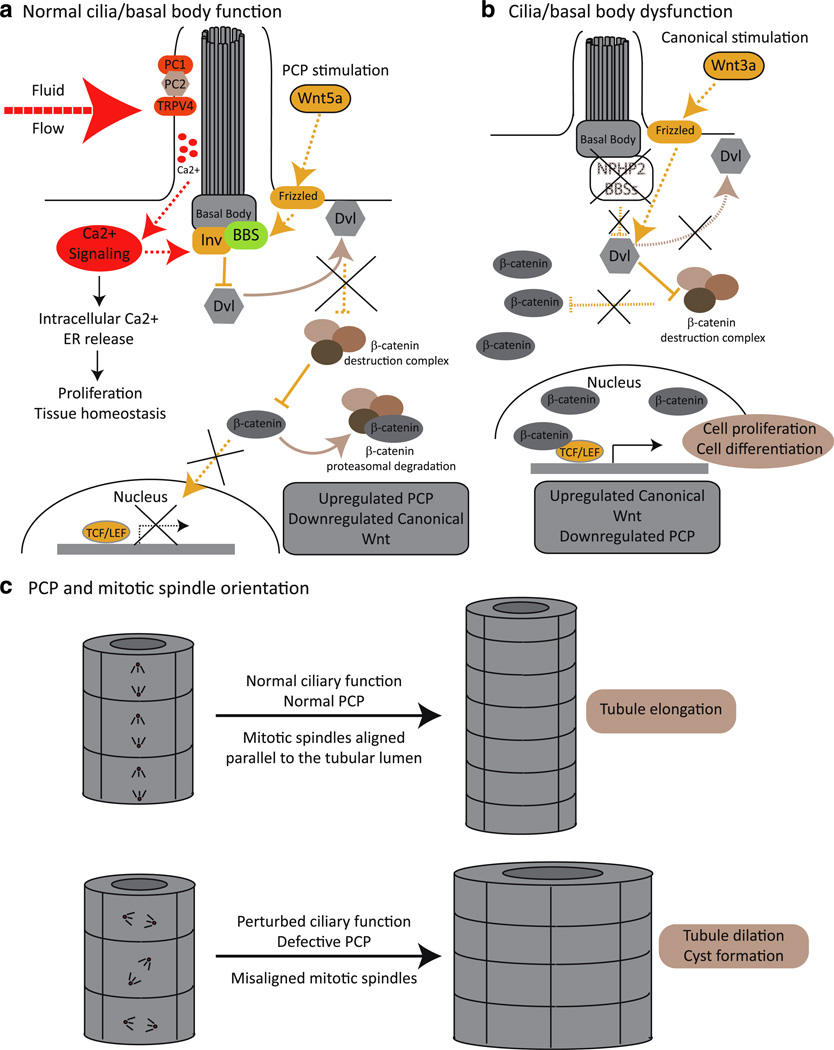
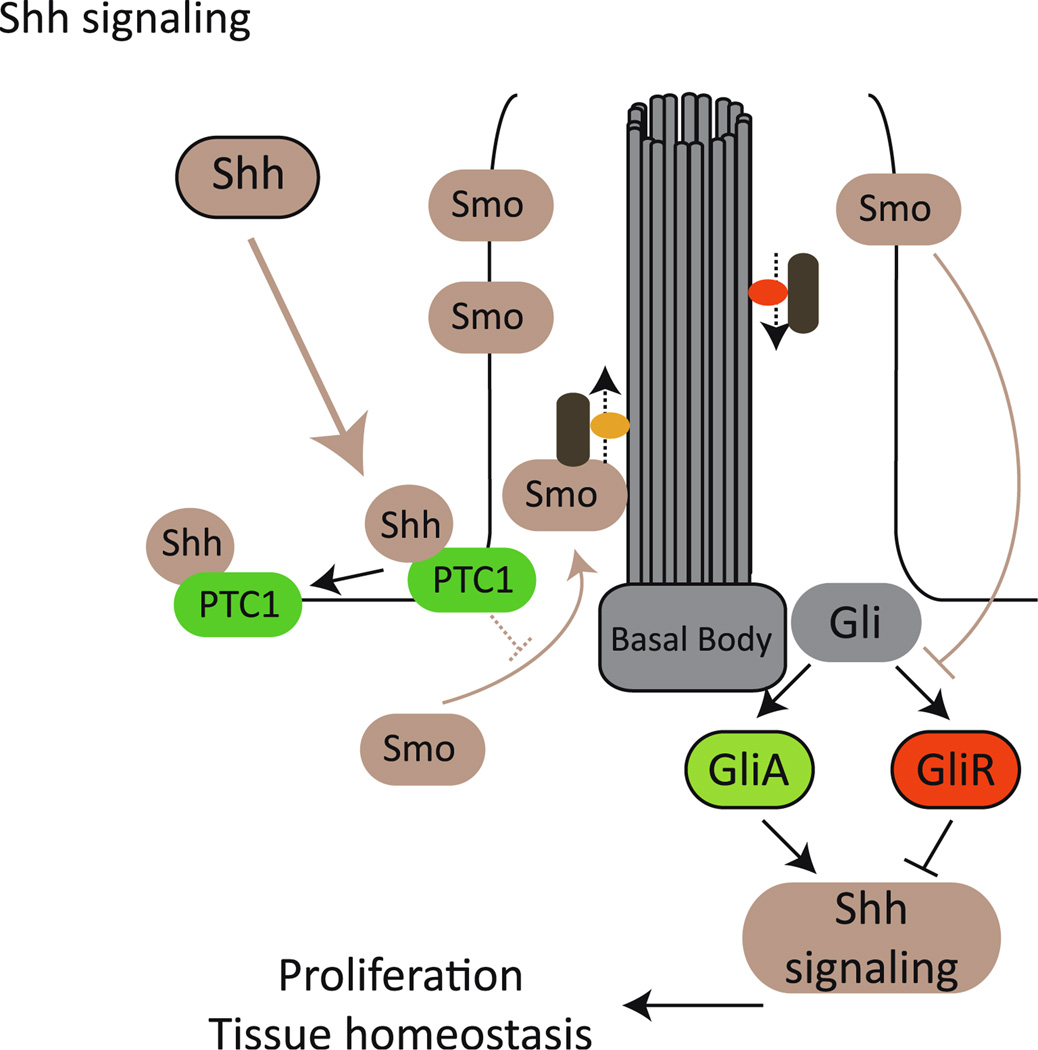
Ciliary dysfunction has emerged as a common factor underlying the pathogenesis of both syndromic and isolated kidney cystic disease, an observation that has contributed to the unification of human genetic disorders of the cilium, the ciliopathies. Such grouping is underscored by two major observations: the fact that genes encoding ciliary proteins can contribute causal and modifying mutations across several clinically discrete ciliopathies, and the emerging realization that an understanding of the clinical pathology of one ciliopathy can provide valuable insight into the pathomechanism of renal cyst formation elsewhere in the ciliopathy spectrum. In this review, we discuss and attempt to stratify the different lines of proposed cilia-driven mechanisms for cystogenesis, ranging from mechano- and chemo-sensation, to cell shape and polarization, to the transduction of a variety of signaling cascades. We evaluate both common trends and differences across the models and discuss how each proposed mechanism can contribute to the development of novel therapeutic paradigms.
Figures
References
-
- Beales PL, Parfrey PS, Katsanis N. The Bardet-Biedl and Alstrom Syndromes. In: Maher E, Saggar-Malik A, editors. Genetics of renal disease. Oxford: Oxford University Press; 2004. pp. 361–398.
-
- Hildebrandt F, Otto E. Cilia and centrosomes: a unifying pathogenic concept for cystic kidney disease? Nat Rev Genet. 2005;6:928–940. - PubMed
-
- Watnick T, Germino G. From cilia to cyst. Nat Genet. 2003;34:355–356. - PubMed
-
- Badano JL, Mitsuma N, Beales PL, Katsanis N. The ciliopathies: an emergin class of human genetic disorders. Annu Rev Genomics Hum Genet. 2006;7:125–148. - PubMed
-
- Fliegauf M, Benzing T, Omran H. When cilia go bad: cilia defects and ciliopathies. Nat Rev Mol Cell Biol. 2007;8:880–893. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
