

Molecular defects in human carbamoy phosphate synthetase I: mutational spectrum, diagnostic and protein structure considerations
- PMID: 21120950
- PMCID: PMC4861085
- DOI: 10.1002/humu.21406
Molecular defects in human carbamoy phosphate synthetase I: mutational spectrum, diagnostic and protein structure considerations
Abstract
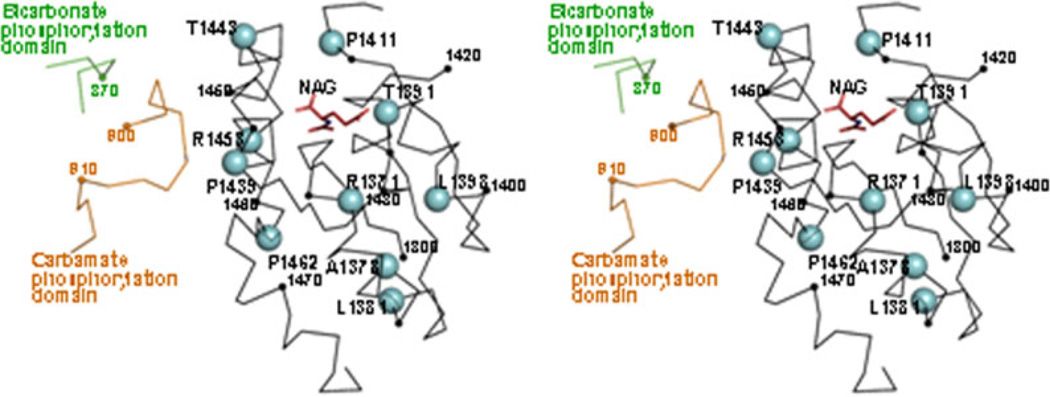
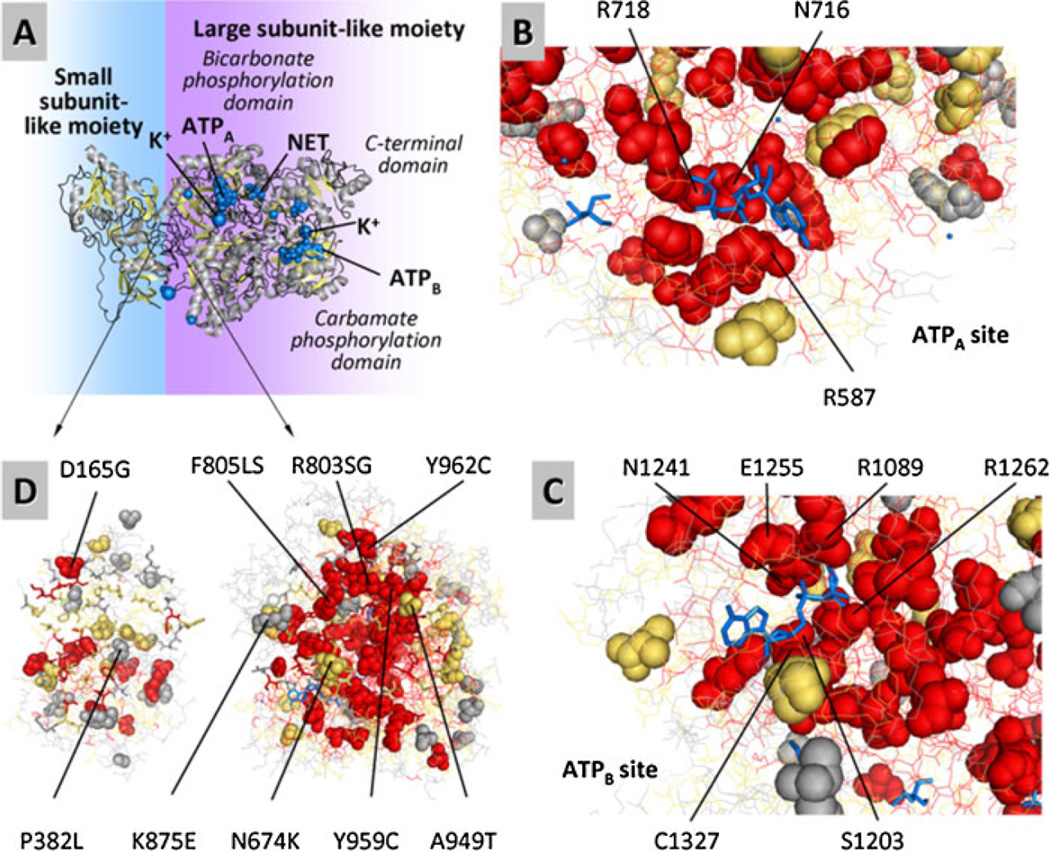
Deficiency of carbamoyl phosphate synthetase I (CPSI) results in hyperammonemia ranging from neonatally lethal to environmentally induced adult-onset disease. Over 24 years, analysis of tissue and DNA samples from 205 unrelated individuals diagnosed with CPSI deficiency (CPSID) detected 192 unique CPS1 gene changes, of which 130 are reported here for the first time. Pooled with the already reported mutations, they constitute a total of 222 changes, including 136 missense, 15 nonsense, 50 changes of other types resulting in enzyme truncation, and 21 other changes causing in-frame alterations. Only ∼10% of the mutations recur in unrelated families, predominantly affecting CpG dinucleotides, further complicating the diagnosis because of the "private" nature of such mutations. Missense changes are unevenly distributed along the gene, highlighting the existence of CPSI regions having greater functional importance than other regions. We exploit the crystal structure of the CPSI allosteric domain to rationalize the effects of mutations affecting it. Comparative modeling is used to create a structural model for the remainder of the enzyme. Missense changes are found to directly correlate, respectively, with the one-residue evolutionary importance and inversely correlate with solvent accessibility of the mutated residue. This is the first large-scale report of CPS1 mutations spanning a wide variety of molecular defects highlighting important regions in this protein.
© 2011 Wiley-Liss, Inc.
Figures



References
-
- Ahuja V, Powers-Lee SG. Human carbamoyl-phosphate synthetase: insight into N-acetylglutamate interaction and the functional effects of a common single nucleotide polymorphism. J Inherit Metab Dis. 2008;31:481–491. - PubMed
-
- Alonso E, Cervera J, Garcia-Espana A, Bendala E, Rubio V. Oxidative inactivation of carbamoyl phosphate synthetase (ammonia). Mechanism and sites of oxidation, degradation of the oxidized enzyme, and inactivation by glycerol, EDTA, and thiol protecting agents. J Biol Chem. 1992;267:4524–4532. - PubMed
-
- Alonso E, Rubio V. Affinity cleavage of carbamoyl-phosphate synthetase I localizes regions of the enzyme interacting with the molecule of ATP that phosphorylates carbamate. Eur J Biochem. 1995;229:377–384. - PubMed
-
- Aoshima T, Kajita M, Sekido Y, Kikuchi S, Yasuda I, Saheki T, Watanabe K, Shimokata K, Niwa T. Novel mutations (H337R and 238-362del) in the CPS1 gene cause carbamoyl phosphate synthetase I deficiency. Hum Hered. 2001a;52:99–101. - PubMed
-
- Aoshima T, Kajita M, Sekido Y, Mimura S, Itakura A, Yasuda I, Saheki T, Watanabe K, Shimokata K, Niwa T. Carbamoyl phosphate synthetase I deficiency: molecular genetic findings and prenatal diagnosis. Prenat Diagn. 2001b;21:634–637. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
