

Inhibition of RIP2's tyrosine kinase activity limits NOD2-driven cytokine responses
- PMID: 21123652
- PMCID: PMC2994040
- DOI: 10.1101/gad.1964410
Inhibition of RIP2's tyrosine kinase activity limits NOD2-driven cytokine responses
Abstract
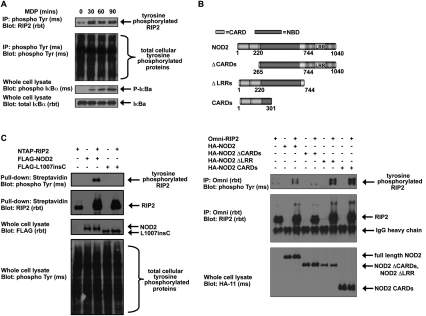
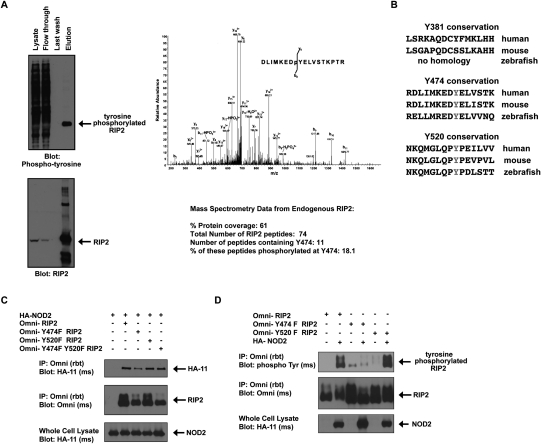
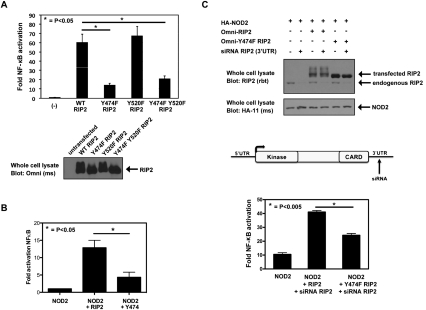
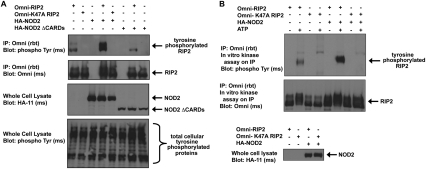
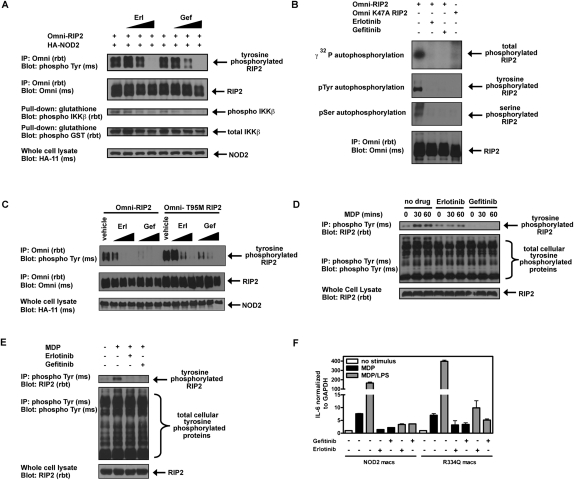
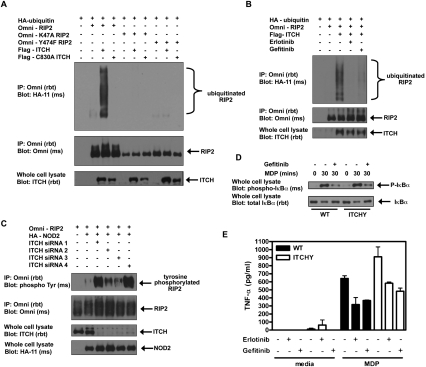
Upon intracellular bacterial exposure, the Crohn's disease and sarcoidosis susceptibility protein NOD2 (nucleotide oligomerization domain protein 2) binds to the protein kinase RIP2 (receptor-interacting protein 2) to coordinate NF-κB (nuclear factor κ B)-mediated cytokine responses. While RIP2 clearly has kinase activity, the function of its kinase domain has been enigmatic. Although originally classified as a serine-threonine kinase based on homology scans, we find that RIP2 also has tyrosine kinase activity. RIP2 undergoes autophosphorylation on Tyr 474 (Y474). This phosphorylation event is necessary for effective NOD2 signaling and does not occur in the presence of the most common Crohn's disease-associated NOD2 allele. Given this tyrosine kinase activity, a small-molecule inhibitor screen designed to identify pharmacologic agents that inhibit RIP2's tyrosine kinase activity was performed. At nanomolar concentrations, the EGFR (epidermal growth factor receptor) tyrosine kinase inhibitors gefitinib (Iressa) and erlotinib (Tarceva) were found to inhibit both RIP2 tyrosine phosphorylation and MDP (muramyl dipeptide)-induced cytokine release in a variety of NOD2 hyperactivation states. This effect is specific for RIP2 and does not depend on EGFR. The finding that RIP2 has tyrosine kinase activity and the finding that gefitinib and erlotinib, two agents already used clinically for cancer chemotherapy, can inhibit this activity suggest that RIP2's tyrosine kinase activity could be targeted specifically in the treatment of inflammatory diseases.
Figures
References
-
- Abbott DW, Wilkins A, Asara JM, Cantley LC 2004. The Crohn's disease protein, NOD2, requires RIP2 in order to induce ubiquitinylation of a novel site on NEMO. Curr Biol 14: 2217–2227 - PubMed
-
- Argast GM, Fausto N, Campbell JS 2005. Inhibition of RIP2/RIck/CARDIAK activity by pyridinyl imidazole inhibitors of p38 MAPK. Mol Cell Biochem 268: 129–140 - PubMed
-
- Bertrand MJ, Doiron K, Labbe K, Korneluk RG, Barker PA, Saleh M 2009. Cellular inhibitors of apoptosis cIAP1 and cIAP2 are required for innate immunity signaling by the pattern recognition receptors NOD1 and NOD2. Immunity 30: 789–801 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous