

Molecular characterization of Borrelia persica, the agent of tick borne relapsing fever in Israel and the Palestinian Authority
- PMID: 21124792
- PMCID: PMC2991353
- DOI: 10.1371/journal.pone.0014105
Molecular characterization of Borrelia persica, the agent of tick borne relapsing fever in Israel and the Palestinian Authority
Abstract
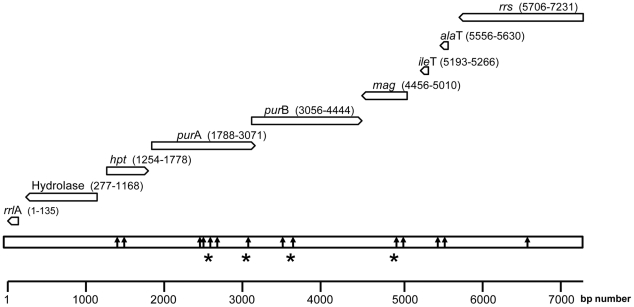
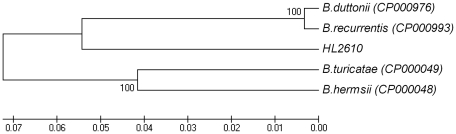
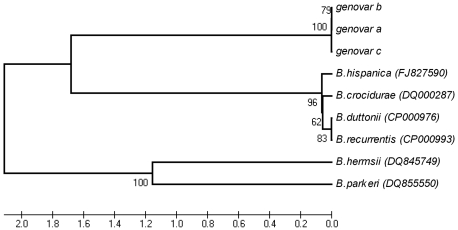
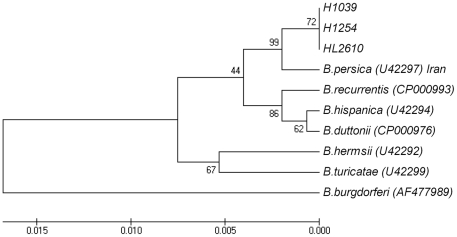
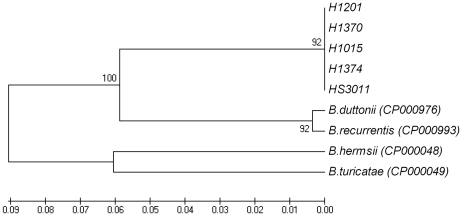
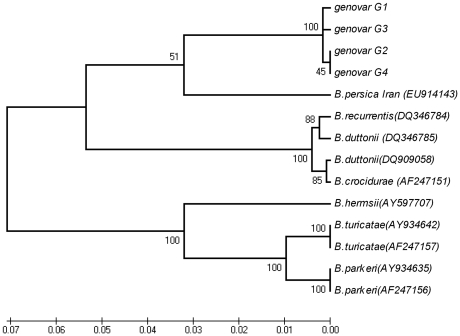
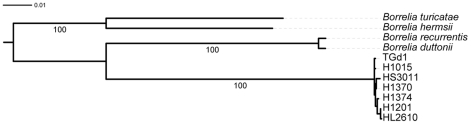
The identification of the Tick Borne Relapsing Fever (TBRF) agent in Israel and the Palestinian Authority relies on the morphology and the association of Borrelia persica with its vector Ornithodoros tholozani. Molecular based data on B. persica are very scarce as the organism is still non-cultivable. In this study, we were able to sequence three complete 16S rRNA genes, 12 partial flaB genes, 18 partial glpQ genes, 16 rrs-ileT intergenic spacers (IGS) from nine ticks and ten human blood samples originating from the West Bank and Israel. In one sample we sequenced 7231 contiguous base pairs that covered completely the region from the 5'end of the 16S rRNA gene to the 5'end of the 23S rRNA gene comprising the whole 16S rRNA (rrs), and the following genes: Ala tRNA (alaT), Ile tRNA (ileT), adenylosuccinate lyase (purB), adenylosuccinate synthetase (purA), methylpurine-DNA glycosylase (mag), hypoxanthine-guanine phosphoribosyltransferase (hpt), an hydrolase (HAD superfamily) and a 135 bp 5' fragment of the 23S rRNA (rrlA) genes. Phylogenic sequence analysis defined all the Borrelia isolates from O. tholozani and from human TBRF cases in Israel and the West Bank as B. persica that clustered between the African and the New World TBRF species. Gene organization of the intergenic spacer between the 16S rRNA and the 23S rRNA was similar to that of other TBRF Borrelia species and different from the Lyme disease Borrelia species. Variants of B. persica were found among the different genes of the different isolates even in the same sampling area.
Conflict of interest statement
Figures
References
-
- Cutler SJ. Relapsing fever–a forgotten disease revealed. J Appl Microbiol. 2010;108:1115–1122. - PubMed
-
- Assous MV, Wilamowski A. Relapsing fever borreliosis in Eurasia- forgotten, but certainly not gone. Clin Microbiol Infect. 2009;15:407–414. - PubMed
-
- Halperin T, Orr N, Cohen R, Hasin T, Davidovitch N, et al. Detection of relapsing fever in human blood samples from Israel using PCR targeting the glycerophosphodiester phosphodiesterase (GlpQ) gene. Acta Tropica. 2006;98:189–195. - PubMed
-
- Rodhain F. Borrelia et fievre recurrentes aspects epidemiologique actuells. Bull Inst Pasteur. 1976;74:173–218.
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
