

Comparative genomics of the bacterial genus Listeria: Genome evolution is characterized by limited gene acquisition and limited gene loss
- PMID: 21126366
- PMCID: PMC3019230
- DOI: 10.1186/1471-2164-11-688
Comparative genomics of the bacterial genus Listeria: Genome evolution is characterized by limited gene acquisition and limited gene loss
Abstract
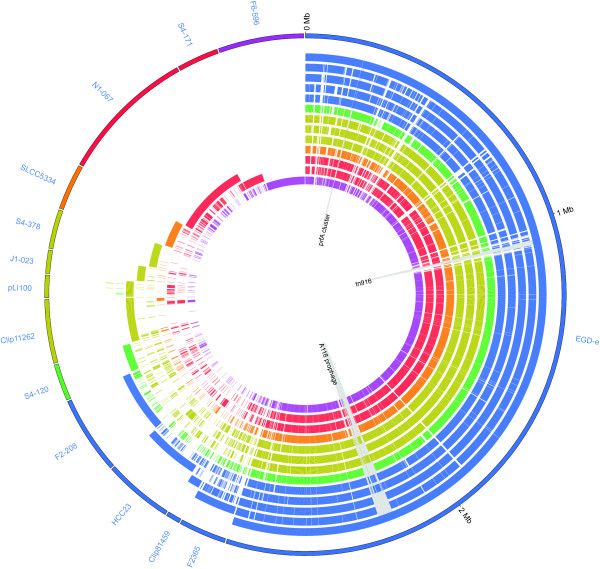
Background: The bacterial genus Listeria contains pathogenic and non-pathogenic species, including the pathogens L. monocytogenes and L. ivanovii, both of which carry homologous virulence gene clusters such as the prfA cluster and clusters of internalin genes. Initial evidence for multiple deletions of the prfA cluster during the evolution of Listeria indicates that this genus provides an interesting model for studying the evolution of virulence and also presents practical challenges with regard to definition of pathogenic strains.
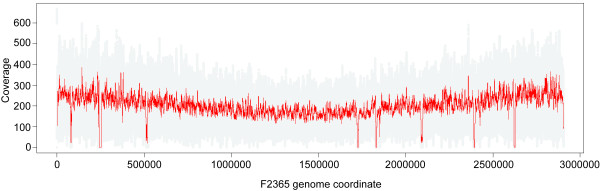
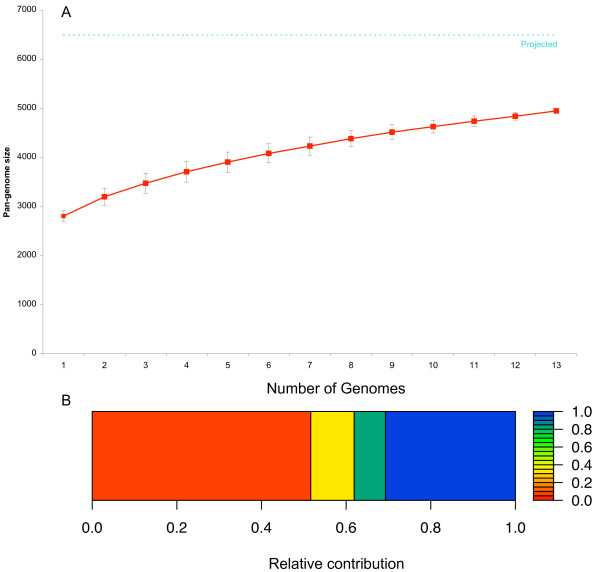
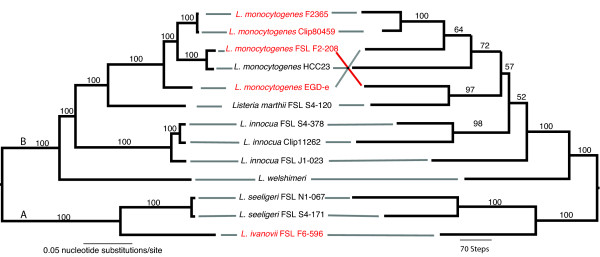
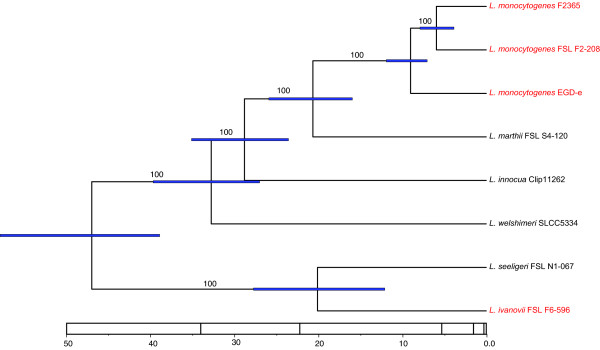
Results: To better understand genome evolution and evolution of virulence characteristics in Listeria, we used a next generation sequencing approach to generate draft genomes for seven strains representing Listeria species or clades for which genome sequences were not available. Comparative analyses of these draft genomes and six publicly available genomes, which together represent the main Listeria species, showed evidence for (i) a pangenome with 2,032 core and 2,918 accessory genes identified to date, (ii) a critical role of gene loss events in transition of Listeria species from facultative pathogen to saprotroph, even though a consistent pattern of gene loss seemed to be absent, and a number of isolates representing non-pathogenic species still carried some virulence associated genes, and (iii) divergence of modern pathogenic and non-pathogenic Listeria species and strains, most likely circa 47 million years ago, from a pathogenic common ancestor that contained key virulence genes.
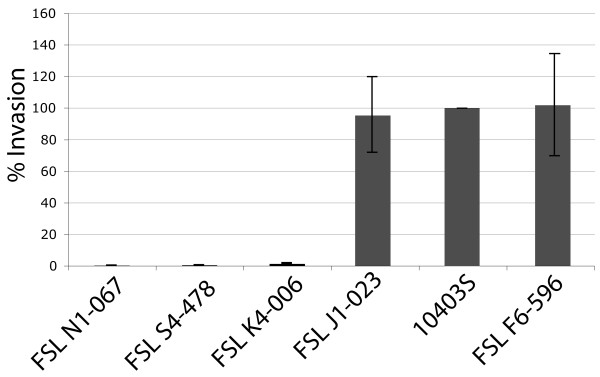
Conclusions: Genome evolution in Listeria involved limited gene loss and acquisition as supported by (i) a relatively high coverage of the predicted pan-genome by the observed pan-genome, (ii) conserved genome size (between 2.8 and 3.2 Mb), and (iii) a highly syntenic genome. Limited gene loss in Listeria did include loss of virulence associated genes, likely associated with multiple transitions to a saprotrophic lifestyle. The genus Listeria thus provides an example of a group of bacteria that appears to evolve through a loss of virulence rather than acquisition of virulence characteristics. While Listeria includes a number of species-like clades, many of these putative species include clades or strains with atypical virulence associated characteristics. This information will allow for the development of genetic and genomic criteria for pathogenic strains, including development of assays that specifically detect pathogenic Listeria strains.
Figures
References
-
- Graves LM, Helsel LO, Steigerwalt AG, Morey RE, Daneshvar MI, Roof SE, Orsi RH, Fortes ED, Millilo SR, den Bakker HC, Wiedmann M, Swaminathan B, Sauders BD. Listeria marthii sp. nov., isolated from the natural environment, Finger Lakes National Forest. Int J Syst Evol Microbiol. 2010;60:1280–1288. doi: 10.1099/ijs.0.014118-0. - DOI - PubMed
-
- Stuart S, Welshimer H. Taxonomic reexamination of Listeria Pirie and transfer of Listeria grayi and Listeria murrayi to a new genus, Murraya. Int J Syst Evol Microbiol. 1974;24:177–185.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
