

Atrial fibrillation induces myocardial fibrosis through angiotensin II type 1 receptor-specific Arkadia-mediated downregulation of Smad7
- PMID: 21127293
- PMCID: PMC3035429
- DOI: 10.1161/CIRCRESAHA.110.234369
Atrial fibrillation induces myocardial fibrosis through angiotensin II type 1 receptor-specific Arkadia-mediated downregulation of Smad7
Abstract
Rationale: Tachycardia-induced atrial fibrosis is a hallmark of structural remodeling of atrial fibrillation (AF). The molecular mechanisms underlying the AF-induced atrial fibrosis remain unclear.
Objective: To determine the role of angiotensin II (Ang II)/Ang II type 1 (AT(1)) receptor-coupled transforming growth factor (TGF)-β(1)/Smad signaling pathway in the AF-induced atrial fibrosis.
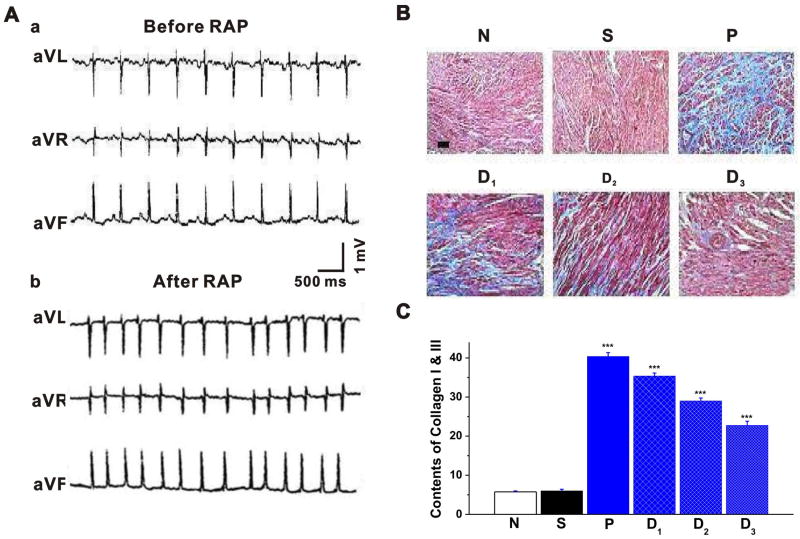
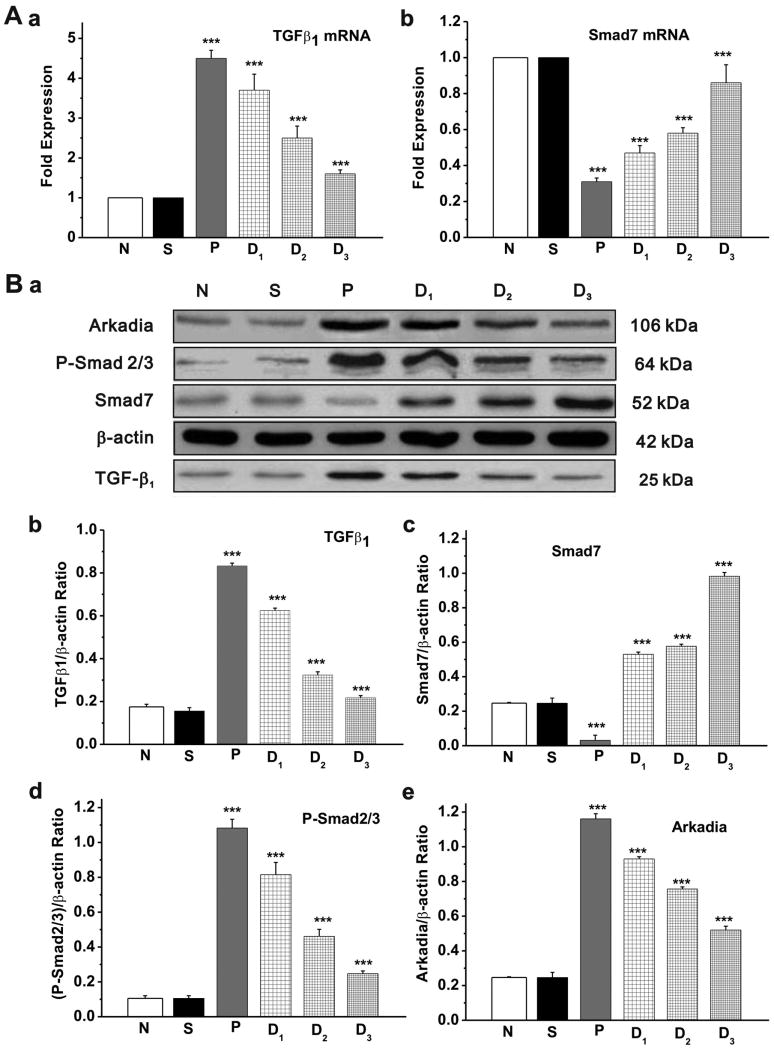
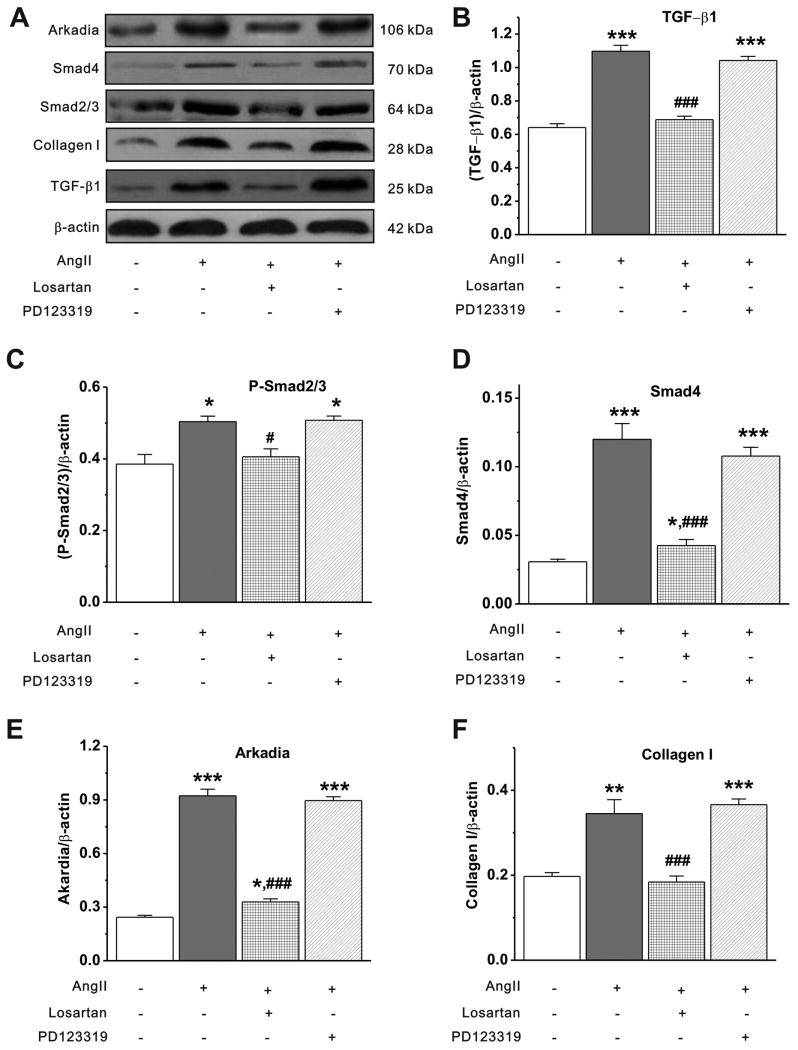
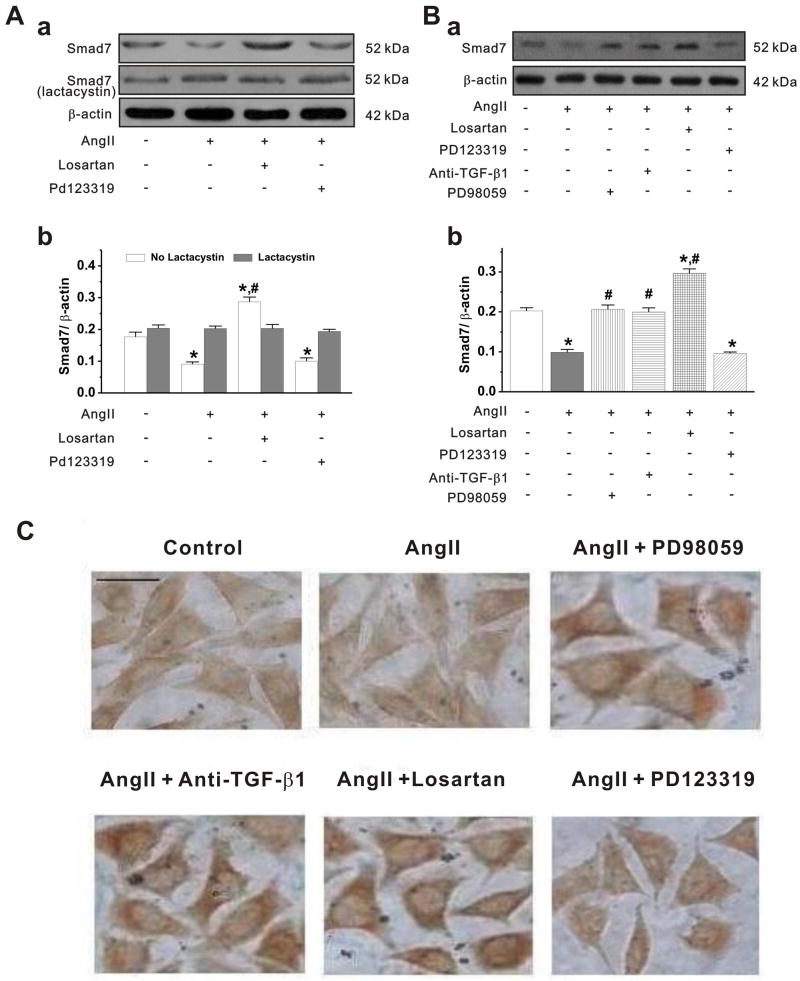
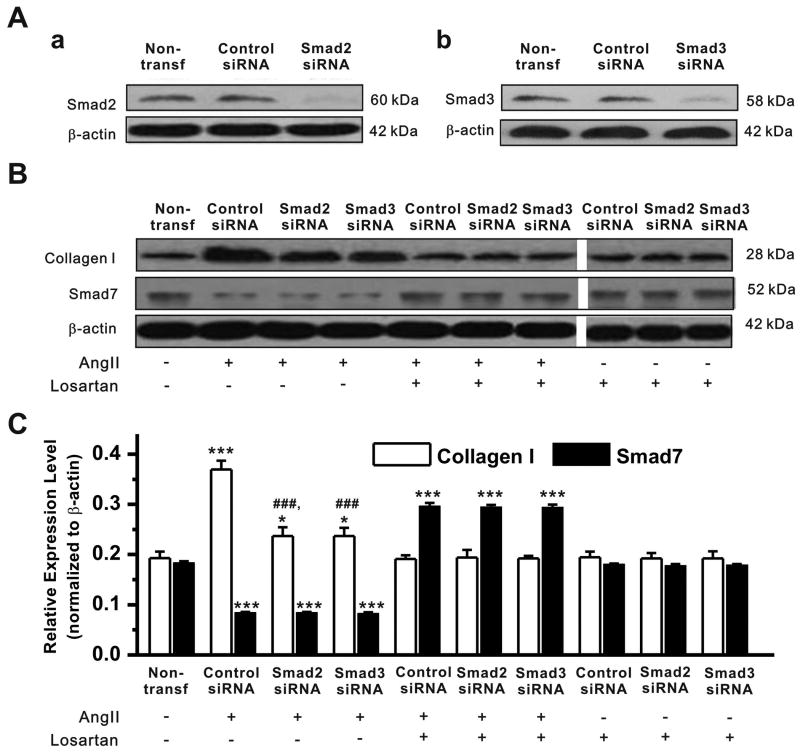
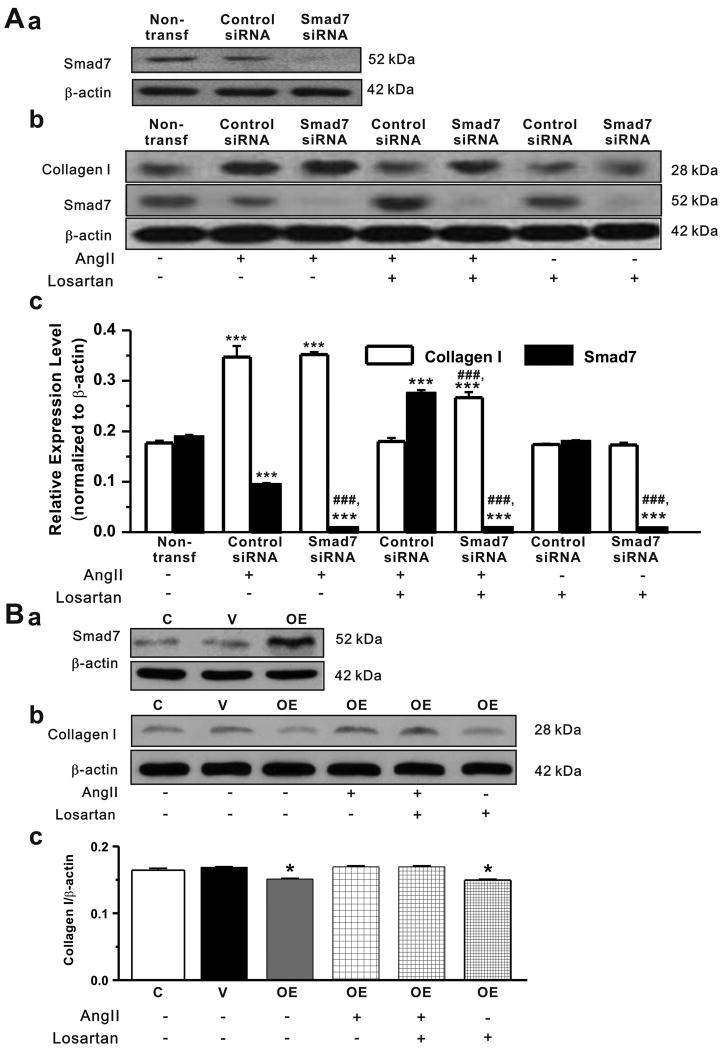
Methods and results: Rapid atrial pacing (1000 ppm) was applied to the left atrium of rabbit heart to induce atrial fibrillation and fibrosis. Quantitative PCR and Western blot analysis revealed that rapid atrial pacing caused a marked increase in the expression of Ang II, TGF-β(1), phosphorylated Smad2/3 (P-Smad2/3), Arkadia, and hydroxyproline synthesis. However, the expression of Smad7, a key endogenous antagonist of the TGF-β(1)/Smad-mediated fibrosis, was significantly decreased. These changes were dose-dependently reversed by AT(1) receptor antagonist losartan, implicating the involvement of AF-induced release of Ang II and activation of AT(1) receptor-specific pathway. In the adult rabbit cardiac fibroblasts, Ang II increased the expression of TGF-β(1), P-Smad2/3, Smad4, Arkadia, and collagen I synthesis and significantly reduced Smad7 expression. These effects of Ang II were reversed by losartan but not by the AT(2) antagonist (PD123319). In addition, extracellular signal-regulated kinase inhibitor and anti-TGF-β(1) antibody also blocked the Ang II-induced downregulation of Smad7. Silencing of Smad7 gene by small interfering RNA abolished the antagonism of losartan on the fibrogenic effects of Ang II on cardiac fibroblasts, whereas overexpression of Smad7 blocked Ang II-induced increase in collagen I synthesis.
Conclusions: Ang II/AT(1) receptor-specific activation of Arkadia-mediated poly-ubiquitination and degradation of Smad7 may decrease the inhibitory feedback regulation of TGF-β(1)/Smad signaling and serves as a key mechanism for AF-induced atrial fibrosis.
Figures
Similar articles
-
Rapid atrial pacing induces myocardial fibrosis by down-regulating Smad7 via microRNA-21 in rabbit.Heart Vessels. 2016 Oct;31(10):1696-708. doi: 10.1007/s00380-016-0808-z. Epub 2016 Mar 11. Heart Vessels. 2016. PMID: 26968995 Free PMC article.
-
Angiotensin II upregulates Kv1.5 expression through ROS-dependent transforming growth factor-beta1 and extracellular signal-regulated kinase 1/2 signalings in neonatal rat atrial myocytes.Biochem Biophys Res Commun. 2014 Nov 21;454(3):410-6. doi: 10.1016/j.bbrc.2014.10.088. Epub 2014 Oct 24. Biochem Biophys Res Commun. 2014. PMID: 25451261
-
Angiotensin II increases collagen I expression via transforming growth factor-beta1 and extracellular signal-regulated kinase in cardiac fibroblasts.Eur J Pharmacol. 2009 Mar 15;606(1-3):115-20. doi: 10.1016/j.ejphar.2008.12.049. Epub 2009 Jan 15. Eur J Pharmacol. 2009. PMID: 19374881
-
Angiotensin II induces connective tissue growth factor and collagen I expression via transforming growth factor-beta-dependent and -independent Smad pathways: the role of Smad3.Hypertension. 2009 Oct;54(4):877-84. doi: 10.1161/HYPERTENSIONAHA.109.136531. Epub 2009 Aug 10. Hypertension. 2009. PMID: 19667256
-
MFGE8 attenuates Ang-II-induced atrial fibrosis and vulnerability to atrial fibrillation through inhibition of TGF-β1/Smad2/3 pathway.J Mol Cell Cardiol. 2020 Feb;139:164-175. doi: 10.1016/j.yjmcc.2020.01.001. Epub 2020 Jan 18. J Mol Cell Cardiol. 2020. PMID: 31958465
Cited by
-
Mining of Potential Biomarkers and Pathway in Valvular Atrial Fibrillation (VAF) via Systematic Screening of Gene Coexpression Network.Comput Math Methods Med. 2022 Oct 3;2022:3645402. doi: 10.1155/2022/3645402. eCollection 2022. Comput Math Methods Med. 2022. Retraction in: Comput Math Methods Med. 2023 Dec 6;2023:9823815. doi: 10.1155/2023/9823815. PMID: 36226239 Free PMC article. Retracted.
-
Rnf111 has a pivotal role in regulating development of definitive hematopoietic stem and progenitor cells through the Smad2/3-Gcsfr/NO axis in zebrafish.Haematologica. 2025 Feb 1;110(2):385-396. doi: 10.3324/haematol.2024.285438. Haematologica. 2025. PMID: 39363867 Free PMC article.
-
Molecular basis of selective atrial fibrosis due to overexpression of transforming growth factor-β1.Cardiovasc Res. 2013 Sep 1;99(4):769-79. doi: 10.1093/cvr/cvt074. Epub 2013 Apr 23. Cardiovasc Res. 2013. PMID: 23612580 Free PMC article.
-
Involvement of Kv1.5 protein in oxidative vascular endothelial cell injury.PLoS One. 2012;7(11):e49758. doi: 10.1371/journal.pone.0049758. Epub 2012 Nov 21. PLoS One. 2012. PMID: 23185428 Free PMC article.
-
Effects of atorvastatin on atrial remodeling in a rabbit model of atrial fibrillation produced by rapid atrial pacing.BMC Cardiovasc Disord. 2016 Jun 24;16(1):142. doi: 10.1186/s12872-016-0301-8. BMC Cardiovasc Disord. 2016. PMID: 27342818 Free PMC article.
References
-
- Blaauw Y, Crijns HJ. Atrial fibrillation: insights from clinical trials and novel treatment options. J Intern Med. 2007;262:593–614. 1. - PubMed
-
- Burstein B, Nattel S. Atrial fibrosis: mechanisms and clinical relevance in atrial fibrillation. J Am Coll Cardiol. 2008;51:802–809. - PubMed
-
- Wyse DG, Gersh BJ. Atrial fibrillation: a perspective: thinking inside and outside the box. Circulation. 2004;109:3089–3095. - PubMed
-
- Beyerbach DM, Zipes DP. Mortality as an endpoint in atrial fibrillation. Heart Rhythm. 2004;1:B8–18. discussion. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
