

Merged consensus clustering to assess and improve class discovery with microarray data
- PMID: 21129181
- PMCID: PMC3002369
- DOI: 10.1186/1471-2105-11-590
Merged consensus clustering to assess and improve class discovery with microarray data
Abstract
Background: One of the most commonly performed tasks when analysing high throughput gene expression data is to use clustering methods to classify the data into groups. There are a large number of methods available to perform clustering, but it is often unclear which method is best suited to the data and how to quantify the quality of the classifications produced.
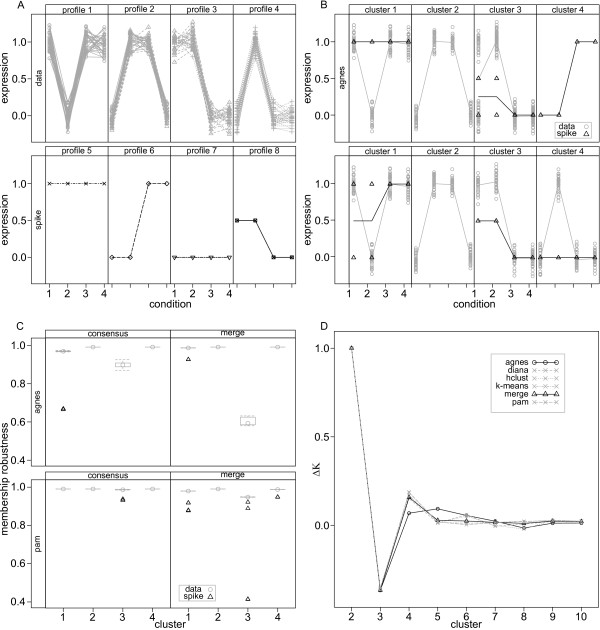
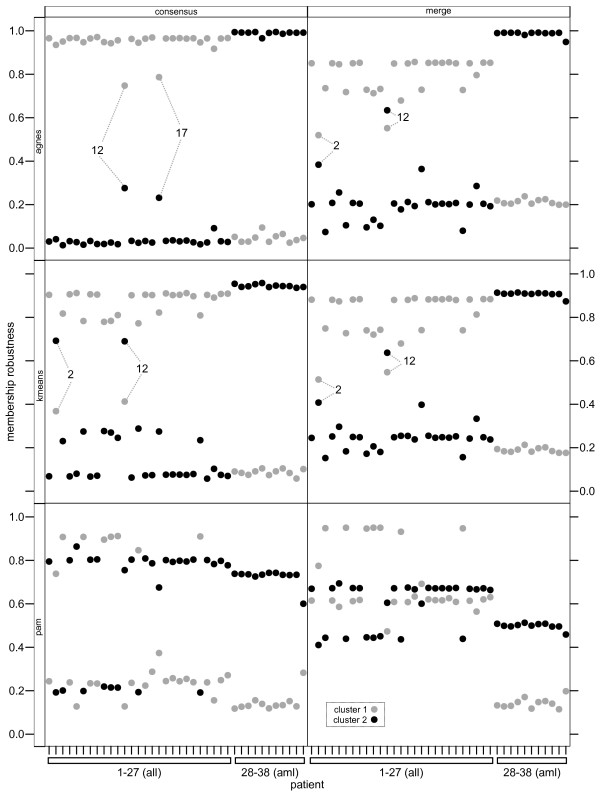
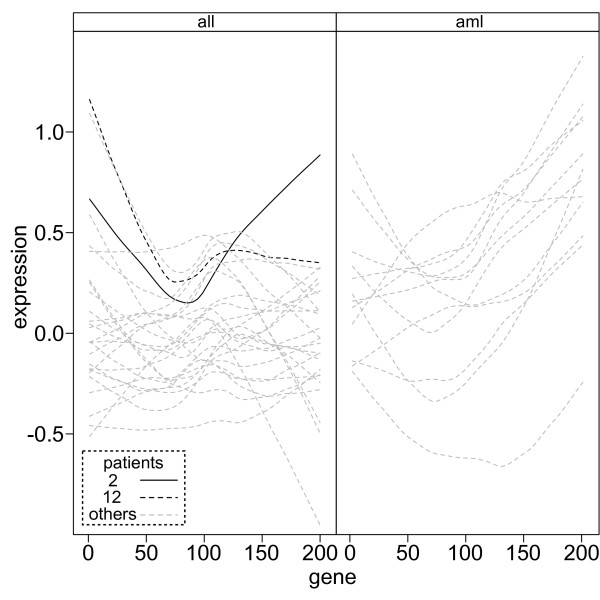
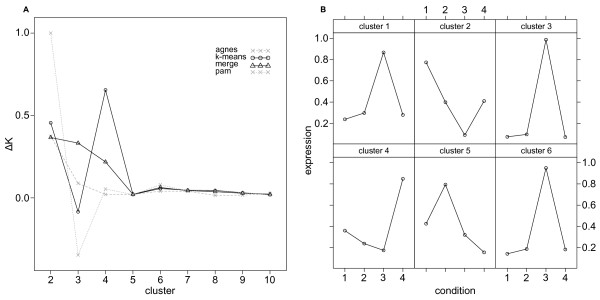
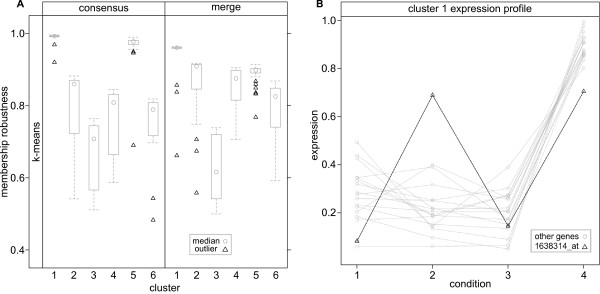
Results: Here we describe an R package containing methods to analyse the consistency of clustering results from any number of different clustering methods using resampling statistics. These methods allow the identification of the the best supported clusters and additionally rank cluster members by their fidelity within the cluster. These metrics allow us to compare the performance of different clustering algorithms under different experimental conditions and to select those that produce the most reliable clustering structures. We show the application of this method to simulated data, canonical gene expression experiments and our own novel analysis of genes involved in the specification of the peripheral nervous system in the fruitfly, Drosophila melanogaster.
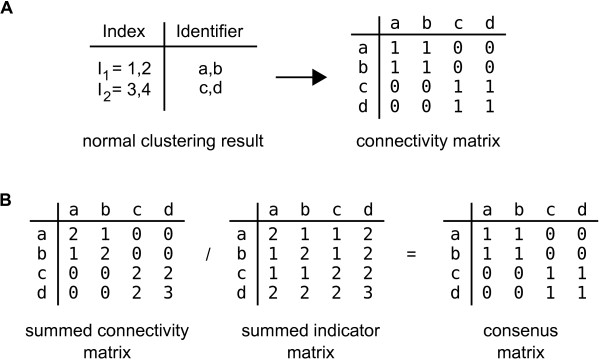
Conclusions: Our package enables users to apply the merged consensus clustering methodology conveniently within the R programming environment, providing both analysis and graphical display functions for exploring clustering approaches. It extends the basic principle of consensus clustering by allowing the merging of results between different methods to provide an averaged clustering robustness. We show that this extension is useful in correcting for the tendency of clustering algorithms to treat outliers differently within datasets. The R package, clusterCons, is freely available at CRAN and sourceforge under the GNU public licence.
Figures
Similar articles
-
Graph-based consensus clustering for class discovery from gene expression data.Bioinformatics. 2007 Nov 1;23(21):2888-96. doi: 10.1093/bioinformatics/btm463. Epub 2007 Sep 14. Bioinformatics. 2007. PMID: 17872912
-
FLAME, a novel fuzzy clustering method for the analysis of DNA microarray data.BMC Bioinformatics. 2007 Jan 4;8:3. doi: 10.1186/1471-2105-8-3. BMC Bioinformatics. 2007. PMID: 17204155 Free PMC article.
-
Clustering of gene expression data: performance and similarity analysis.BMC Bioinformatics. 2006 Dec 12;7 Suppl 4(Suppl 4):S19. doi: 10.1186/1471-2105-7-S4-S19. BMC Bioinformatics. 2006. PMID: 17217511 Free PMC article.
-
Comparing algorithms for clustering of expression data: how to assess gene clusters.Methods Mol Biol. 2009;541:479-509. doi: 10.1007/978-1-59745-243-4_21. Methods Mol Biol. 2009. PMID: 19381534 Review.
-
Unsupervised pattern recognition: an introduction to the whys and wherefores of clustering microarray data.Brief Bioinform. 2005 Dec;6(4):331-43. doi: 10.1093/bib/6.4.331. Brief Bioinform. 2005. PMID: 16420732 Review.
Cited by
-
An alternative to current psychiatric classifications: a psychological landscape hypothesis based on an integrative, dynamical and multidimensional approach.Philos Ethics Humanit Med. 2014 Jul 17;9:12. doi: 10.1186/1747-5341-9-12. Philos Ethics Humanit Med. 2014. PMID: 25033795 Free PMC article.
-
Synaptic Interactome Mining Reveals p140Cap as a New Hub for PSD Proteins Involved in Psychiatric and Neurological Disorders.Front Mol Neurosci. 2017 Jun 30;10:212. doi: 10.3389/fnmol.2017.00212. eCollection 2017. Front Mol Neurosci. 2017. PMID: 28713243 Free PMC article.
-
Is there still a French eating model? A taxonomy of eating behaviors in adults living in the Paris metropolitan area in 2010.PLoS One. 2015 Mar 3;10(3):e0119161. doi: 10.1371/journal.pone.0119161. eCollection 2015. PLoS One. 2015. PMID: 25734543 Free PMC article.
-
Gene expression profiling of CD8+ T cells predicts prognosis in patients with Crohn disease and ulcerative colitis.J Clin Invest. 2011 Oct;121(10):4170-9. doi: 10.1172/JCI59255. Epub 2011 Sep 26. J Clin Invest. 2011. PMID: 21946256 Free PMC article.
-
Dissecting the Shared and Context-Dependent Pathways Mediated by the p140Cap Adaptor Protein in Cancer and in Neurons.Front Cell Dev Biol. 2019 Oct 15;7:222. doi: 10.3389/fcell.2019.00222. eCollection 2019. Front Cell Dev Biol. 2019. PMID: 31681758 Free PMC article.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
