

PSMB8 encoding the β5i proteasome subunit is mutated in joint contractures, muscle atrophy, microcytic anemia, and panniculitis-induced lipodystrophy syndrome
- PMID: 21129723
- PMCID: PMC2997366
- DOI: 10.1016/j.ajhg.2010.10.031
PSMB8 encoding the β5i proteasome subunit is mutated in joint contractures, muscle atrophy, microcytic anemia, and panniculitis-induced lipodystrophy syndrome
Abstract
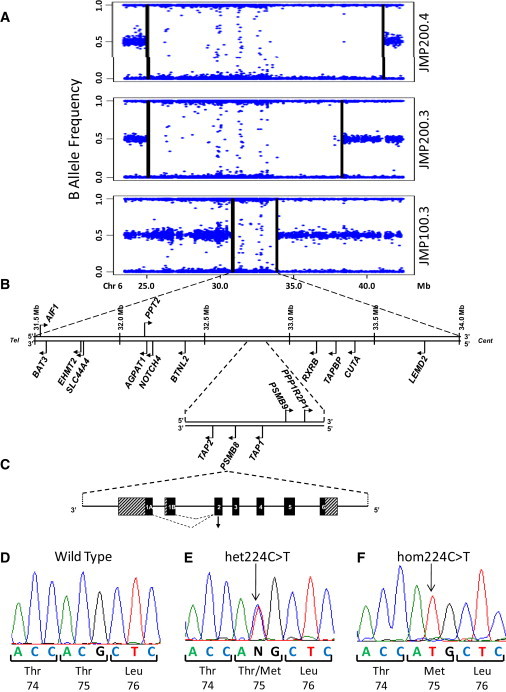
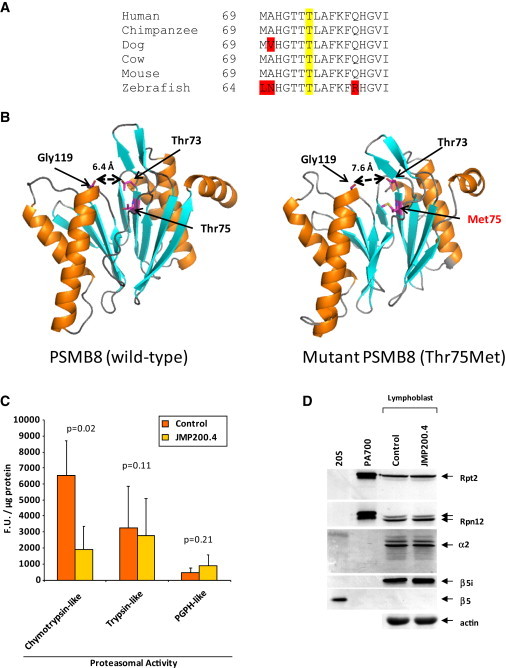
We performed homozygosity mapping in two recently reported pedigrees from Portugal and Mexico with an autosomal-recessive autoinflammatory syndrome characterized by joint contractures, muscle atrophy, microcytic anemia, and panniculitis-induced lipodystrophy (JMP). This revealed only one homozygous region spanning 2.4 Mb (5818 SNPs) on chromosome 6p21 shared by all three affected individuals from both families. We directly sequenced genes involved in immune response located in this critical region, excluding the HLA complex genes. We found a homozygous missense mutation c.224C>T (p.Thr75Met) in the proteasome subunit, beta-type, 8 (PSMB8) gene in affected patients from both pedigrees. The mutation segregated in an autosomal-recessive fashion and was not detected in 275 unrelated ethnically matched healthy subjects. PSMB8 encodes a catalytic subunit of the 20S immunoproteasomes called β5i. Immunoproteasome-mediated proteolysis generates immunogenic epitopes presented by major histocompatibility complex (MHC) class I molecules. Threonine at position 75 is highly conserved and its substitution with methionine disrupts the tertiary structure of PSMB8. As compared to normal lymphoblasts, those from an affected patient showed significantly reduced chymotrypsin-like proteolytic activity mediated by immunoproteasomes. We conclude that mutations in PSMB8 cause JMP syndrome, most probably by affecting MHC class I antigen processing.
Copyright © 2010 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Tanaka M., Miyatani N., Yamada S., Miyashita K., Toyoshima I., Sakuma K., Tanaka K., Yuasa T., Miyatake T., Tsubaki T. Hereditary lipo-muscular atrophy with joint contracture, skin eruptions and hyper-gamma-globulinemia: A new syndrome. Intern. Med. 1993;32:42–45. - PubMed
-
- Yamada S., Toyoshima I., Mori S., Tsubaki T. [Sibling cases with lipodystrophic skin change, muscular atrophy, recurrent skin eruptions, and deformities and contractures of the joints. A possible new clinical entity] Rinsho Shinkeigaku. 1984;24:703–710. - PubMed
-
- Oyanagi K., Sasaki K., Ohama E., Ikuta F., Kawakami A., Miyatani N., Miyatake T., Yamada S. An autopsy case of a syndrome with muscular atrophy, decreased subcutaneous fat, skin eruption and hyper gamma-globulinemia: Peculiar vascular changes and muscle fiber degeneration. Acta Neuropathol. 1987;73:313–319. - PubMed
-
- Horikoshi A., Iwabuchi S., Iizuka Y., Hagiwara T., Amaki I. [A case of partial lipodystrophy with erythema, dactylic deformities, calcification of the basal ganglia, immunological disorders, and low IQ level (author's transl)] Rinsho Shinkeigaku. 1980;20:173–180. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
