

Loss of CHSY1, a secreted FRINGE enzyme, causes syndromic brachydactyly in humans via increased NOTCH signaling
- PMID: 21129727
- PMCID: PMC2997365
- DOI: 10.1016/j.ajhg.2010.11.005
Loss of CHSY1, a secreted FRINGE enzyme, causes syndromic brachydactyly in humans via increased NOTCH signaling
Abstract
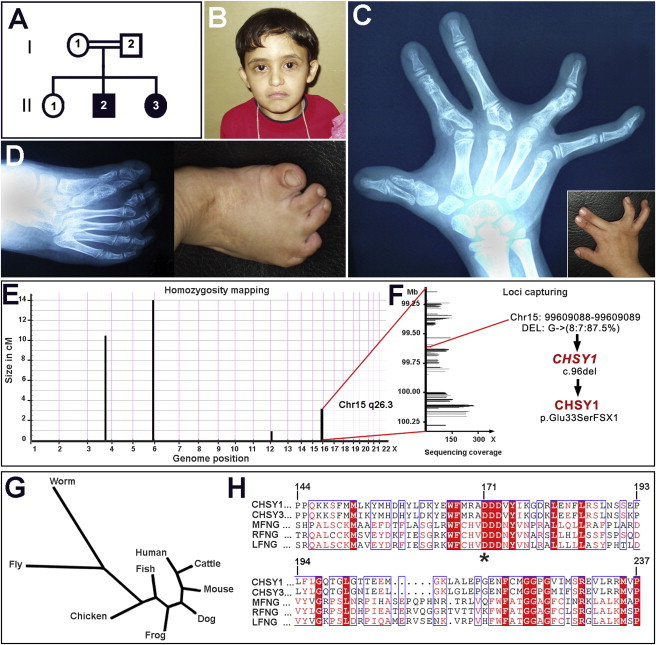
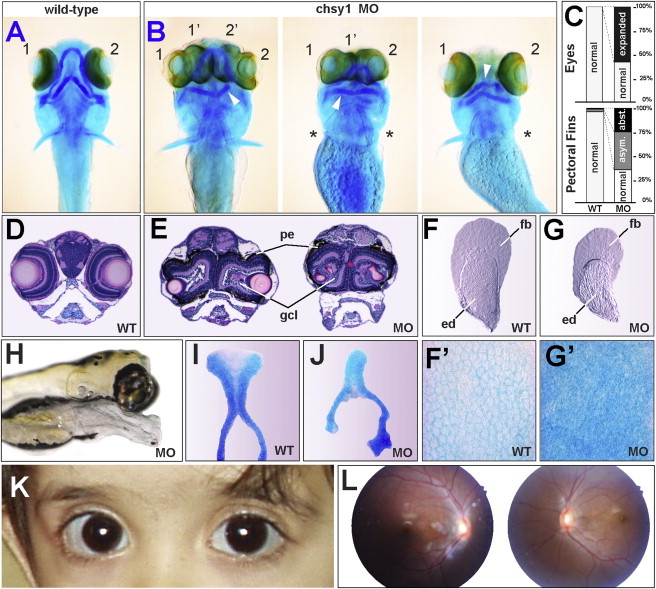
We delineated a syndromic recessive preaxial brachydactyly with partial duplication of proximal phalanges to 16.8 Mb over 4 chromosomes. High-throughput sequencing of all 177 candidate genes detected a truncating frameshift mutation in the gene CHSY1 encoding a chondroitin synthase with a Fringe domain. CHSY1 was secreted from patients' fibroblasts and was required for synthesis of chondroitin sulfate moieties. Noticeably, its absence triggered massive production of JAG1 and subsequent NOTCH activation, which could only be reversed with a wild-type but not a Fringe catalytically dead CHSY1 construct. In vitro, depletion of CHSY1 by RNAi knockdown resulted in enhanced osteogenesis in fetal osteoblasts and remarkable upregulation of JAG2 in glioblastoma cells. In vivo, chsy1 knockdown in zebrafish embryos partially phenocopied the human disorder; it increased NOTCH output and impaired skeletal, pectoral-fin, and retinal development. We conclude that CHSY1 is a secreted FRINGE enzyme required for adjustment of NOTCH signaling throughout human and fish embryogenesis and particularly during limb patterning.
Copyright © 2010 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures



References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
