

Two genetic forms of hyperinsulinemic hypoglycemia caused by dysregulation of glutamate dehydrogenase
- PMID: 21130127
- PMCID: PMC3081417
- DOI: 10.1016/j.neuint.2010.11.017
Two genetic forms of hyperinsulinemic hypoglycemia caused by dysregulation of glutamate dehydrogenase
Abstract
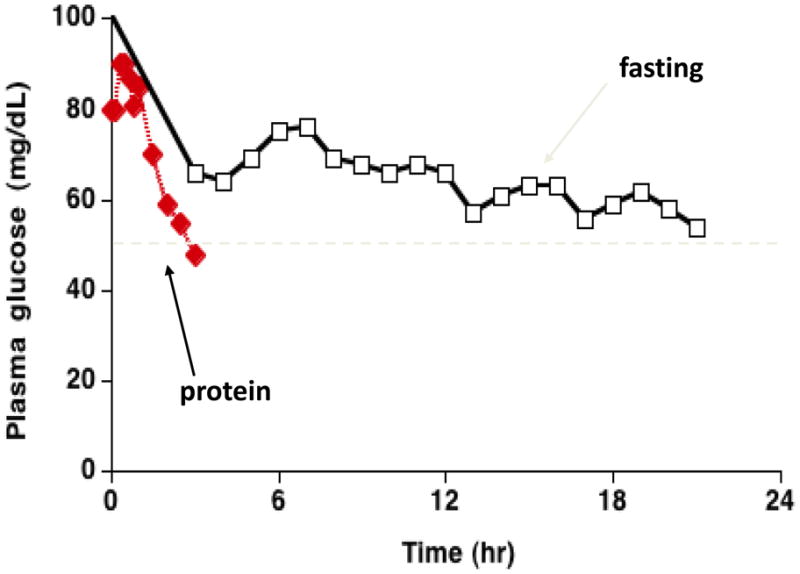
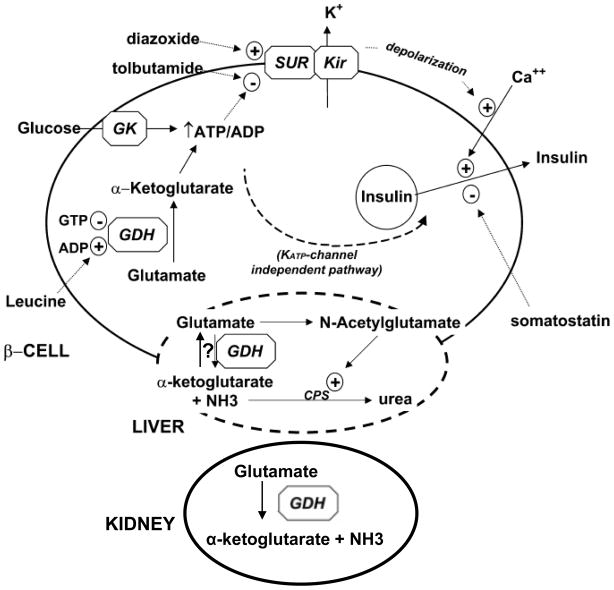
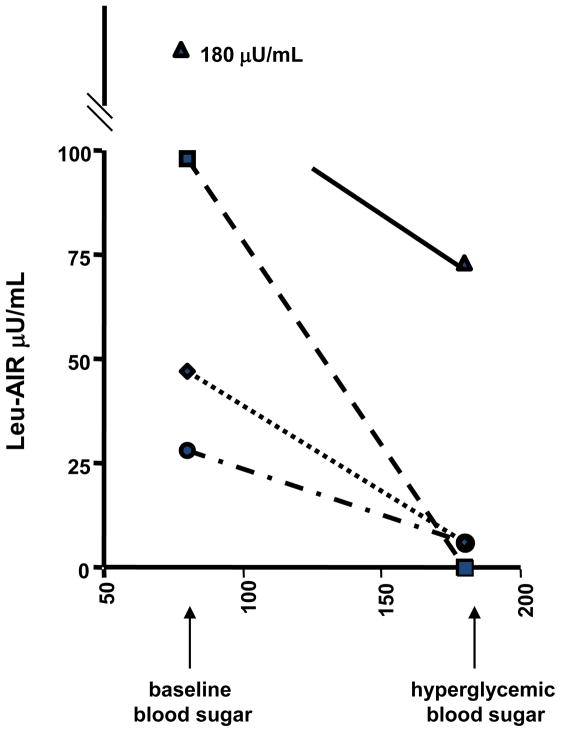
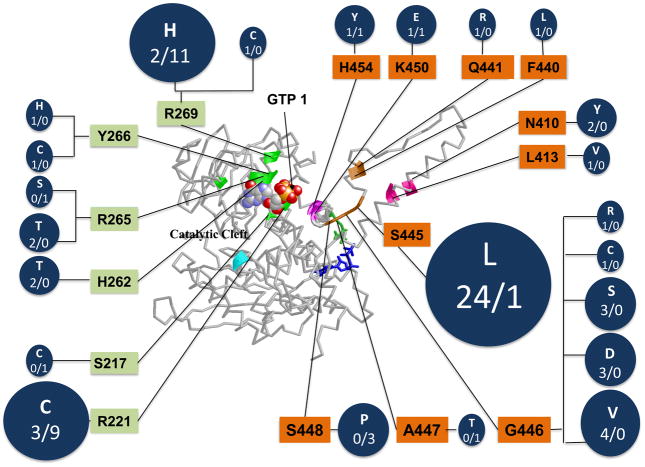
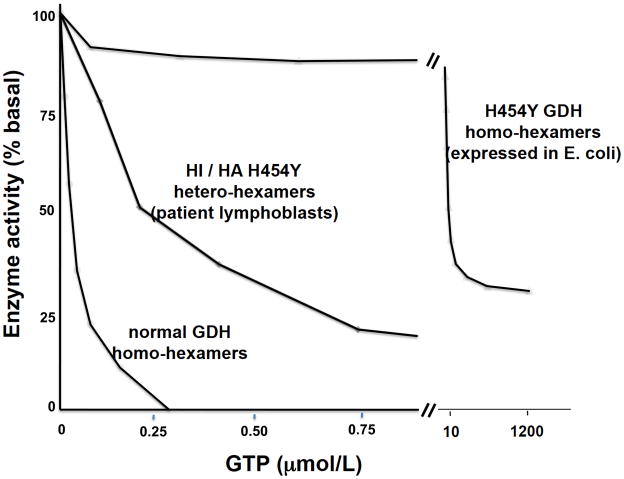
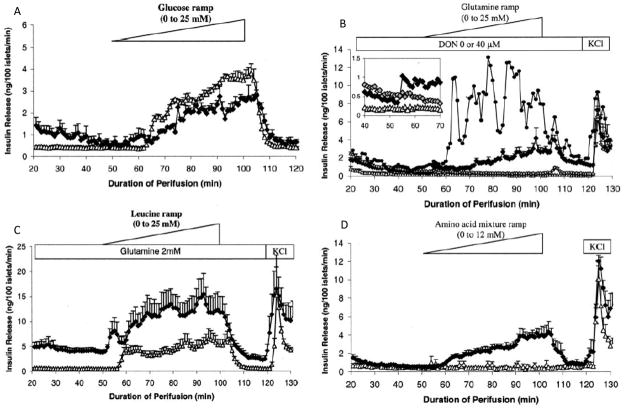
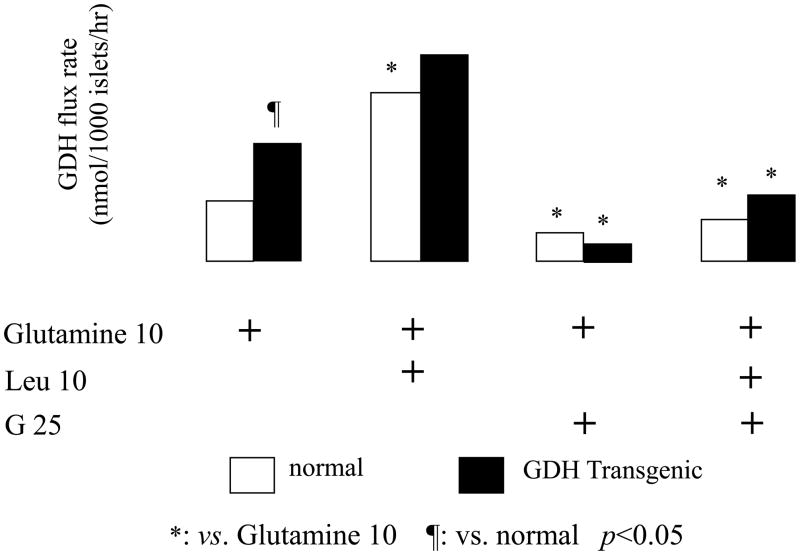
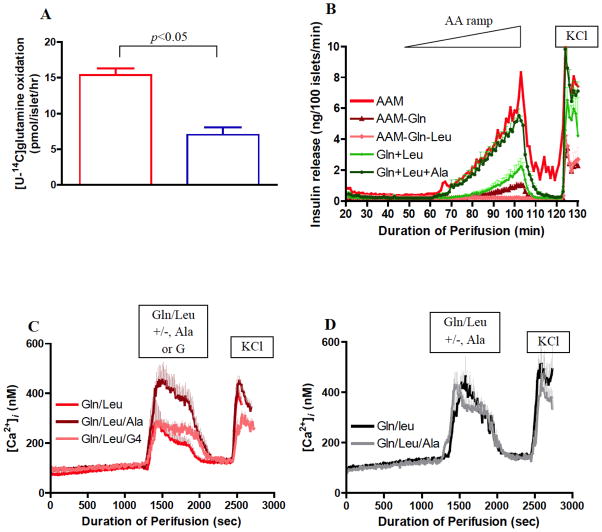
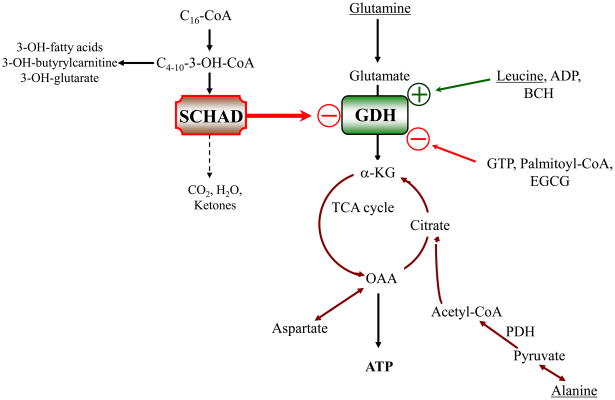
Glutamate dehydrogenase (GDH) has recently been shown to be involved in two genetic disorders of hyperinsulinemic hypoglycemia in children. These include the hyperinsulinism/hyperammonemia syndrome caused by dominant activating mutations of GLUD1 which interfere with inhibitory regulation by GTP and hyperinsulinism due to recessive deficiency of short-chain 3-hydroxy-acyl-CoA dehydrogenase (SCHAD, encoded by HADH1). The clinical manifestations of the abnormalities in pancreatic ß-cell insulin regulation include fasting hypoglycemia, as well as protein-sensitive hypoglycemia. The latter is due to abnormally increased sensitivity of affected children to stimulation of insulin secretion by the amino acid, leucine. In patients with GDH activating mutations, mild hyperammonemia occurs in both the basal and protein-fed state, possibly due to increased renal ammoniagenesis. Some patients with GDH activating mutations appear to be at unusual risk of developmental delay and generalized epilepsy, perhaps reflecting consequences of increased GDH activity in the brain. Studies of these two disorders have been carried out in mouse models to define the mechanisms of insulin dysregulation. In SCHAD deficiency, the activation of GDH is due to loss of a direct inhibitory protein-protein interaction between SCHAD and GDH. These two novel human disorders demonstrate the important role of GDH in insulin regulation and illustrate unexpectedly important reasons for the unusually complex allosteric regulation of GDH.
Copyright © 2010 Elsevier Ltd. All rights reserved.
Figures
Similar articles
-
Identification of the molecular dysfunction caused by glutamate dehydrogenase S445L mutation responsible for hyperinsulinism/hyperammonemia.Hum Mol Genet. 2017 Sep 15;26(18):3453-3465. doi: 10.1093/hmg/ddx213. Hum Mol Genet. 2017. PMID: 28911206
-
Mechanism of hyperinsulinism in short-chain 3-hydroxyacyl-CoA dehydrogenase deficiency involves activation of glutamate dehydrogenase.J Biol Chem. 2010 Oct 8;285(41):31806-18. doi: 10.1074/jbc.M110.123638. Epub 2010 Jul 29. J Biol Chem. 2010. PMID: 20670938 Free PMC article.
-
Regulation of glutamate metabolism and insulin secretion by glutamate dehydrogenase in hypoglycemic children.Am J Clin Nutr. 2009 Sep;90(3):862S-866S. doi: 10.3945/ajcn.2009.27462AA. Epub 2009 Jul 22. Am J Clin Nutr. 2009. PMID: 19625687 Free PMC article. Review.
-
Mitochondrial GTP insensitivity contributes to hypoglycemia in hyperinsulinemia hyperammonemia by inhibiting glucagon release.Diabetes. 2014 Dec;63(12):4218-29. doi: 10.2337/db14-0783. Epub 2014 Jul 14. Diabetes. 2014. PMID: 25024374 Free PMC article.
-
The structure and allosteric regulation of mammalian glutamate dehydrogenase.Arch Biochem Biophys. 2012 Mar 15;519(2):69-80. doi: 10.1016/j.abb.2011.10.015. Epub 2011 Nov 4. Arch Biochem Biophys. 2012. PMID: 22079166 Free PMC article. Review.
Cited by
-
Genetic pathogenesis, diagnosis, and treatment of short-chain 3-hydroxyacyl-coenzyme A dehydrogenase hyperinsulinism.Orphanet J Rare Dis. 2021 Nov 4;16(1):467. doi: 10.1186/s13023-021-02088-6. Orphanet J Rare Dis. 2021. PMID: 34736508 Free PMC article. Review.
-
The odyssey of a young gene: structure-function studies in human glutamate dehydrogenases reveal evolutionary-acquired complex allosteric regulation mechanisms.Neurochem Res. 2014;39(3):471-86. doi: 10.1007/s11064-014-1251-0. Epub 2014 Feb 11. Neurochem Res. 2014. PMID: 24515454 Review.
-
Mechanisms of the amplifying pathway of insulin secretion in the β cell.Pharmacol Ther. 2017 Nov;179:17-30. doi: 10.1016/j.pharmthera.2017.05.003. Epub 2017 May 18. Pharmacol Ther. 2017. PMID: 28527919 Free PMC article. Review.
-
The Pancreatic ß-cell Response to Secretory Demands and Adaption to Stress.Endocrinology. 2021 Nov 1;162(11):bqab173. doi: 10.1210/endocr/bqab173. Endocrinology. 2021. PMID: 34407177 Free PMC article. Review.
-
Allosteric regulation of glutamate dehydrogenase deamination activity.Sci Rep. 2020 Oct 5;10(1):16523. doi: 10.1038/s41598-020-73743-4. Sci Rep. 2020. PMID: 33020580 Free PMC article.
References
-
- Li C, Matter A, Kelly A, et al. Effects of a GTP-insensitive mutation of glutamate dehydrogenase on insulin secretion in transgenic mice. J Biol Chem. 2006;281:15064–72. - PubMed
-
- Stanley CA, Lieu YK, Hsu BY, et al. Hyperinsulinism and hyperammonemia in infants with regulatory mutations of the glutamate dehydrogenase gene. The New England journal of medicine. 1998;338:1352–7. - PubMed
-
- Smith TJ, Stanley CA. Untangling the glutamate dehydrogenase allosteric nightmare. Trends in biochemical sciences. 2008 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01-DK072171/DK/NIDDK NIH HHS/United States
- S10-RR026853/RR/NCRR NIH HHS/United States
- R01-DK 56268/DK/NIDDK NIH HHS/United States
- S10 RR026853/RR/NCRR NIH HHS/United States
- R01 DK072171/DK/NIDDK NIH HHS/United States
- R01 DK053012/DK/NIDDK NIH HHS/United States
- P30 DK019525/DK/NIDDK NIH HHS/United States
- UL1 RR024134/RR/NCRR NIH HHS/United States
- R37 DK056268/DK/NIDDK NIH HHS/United States
- DK 53012/DK/NIDDK NIH HHS/United States
- P30-DK19525/DK/NIDDK NIH HHS/United States
- UL1-RR024134/RR/NCRR NIH HHS/United States
- R01 DK056268/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
