

Novel GNE mutations in two phenotypically distinct HIBM2 patients
- PMID: 21131200
- PMCID: PMC3030125
- DOI: 10.1016/j.nmd.2010.11.002
Novel GNE mutations in two phenotypically distinct HIBM2 patients
Abstract
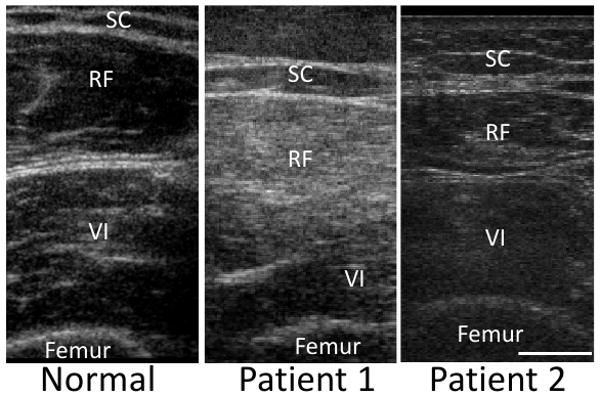
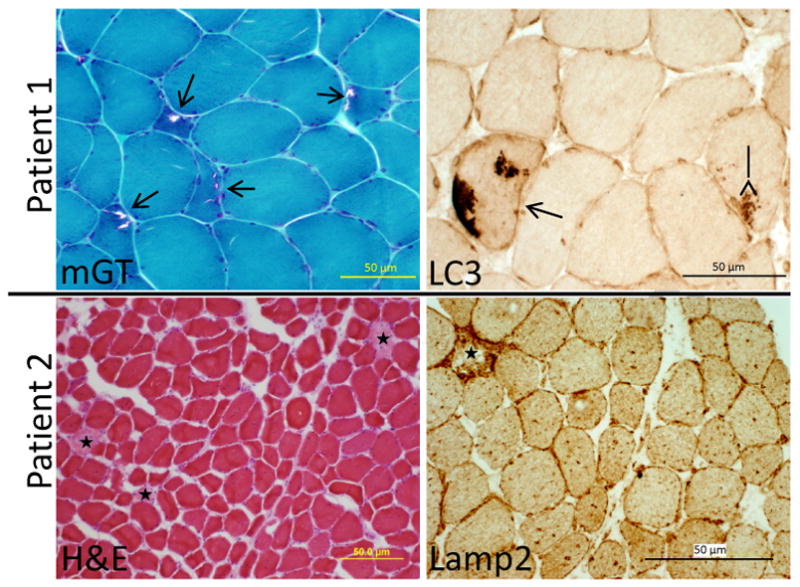
Homozygous mutations in the UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase (GNE) gene cause hereditary inclusion body myopathy type 2 (HIBM2). We describe two unrelated American patients with novel GNE mutations. While one patient followed a typical disease course for HIBM2 with an onset at age 25 and rimmed vacuole pathology on muscle biopsy, the second patient had several features atypical for HIBM2. This patient's onset was at age 55, included distal weakness, quadriceps sparing and respiratory insufficiency. His muscle biopsy showed prominent necrosis without rimmed vacuoles. This study expands the phenotype and illustrates the clinical spectrum of HIBM2 identified in a U.S. based neuromuscular clinic.
Copyright © 2010 Elsevier B.V. All rights reserved.
Figures
References
-
- Eisenberg I, et al. The UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase gene is mutated in recessive hereditary inclusion body myopathy. Nat Genet. 2001;29(1):83–7. - PubMed
-
- Huizing M, et al. Hypoglycosylation of alpha-dystroglycan in patients with hereditary IBM due to GNE mutations. Mol Genet Metab. 2004;81(3):196–202. - PubMed
-
- Salama I, et al. No overall hyposialylation in hereditary inclusion body myopathy myoblasts carrying the homozygous M712T GNE mutation. Biochem Biophys Res Commun. 2005;328(1):221–6. - PubMed
-
- Nishino I, et al. Distal myopathy with rimmed vacuoles is allelic to hereditary inclusion body myopathy. Neurology. 2002;59(11):1689–93. - PubMed
-
- Saechao C, et al. Novel GNE mutations in hereditary inclusion body myopathy patients of non-Middle Eastern descent. Genet Test Mol Biomarkers. 14(2):157–62. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
