

Transcription of functionally related constitutive genes is not coordinated
- PMID: 21131977
- PMCID: PMC3058351
- DOI: 10.1038/nsmb.1934
Transcription of functionally related constitutive genes is not coordinated
Abstract
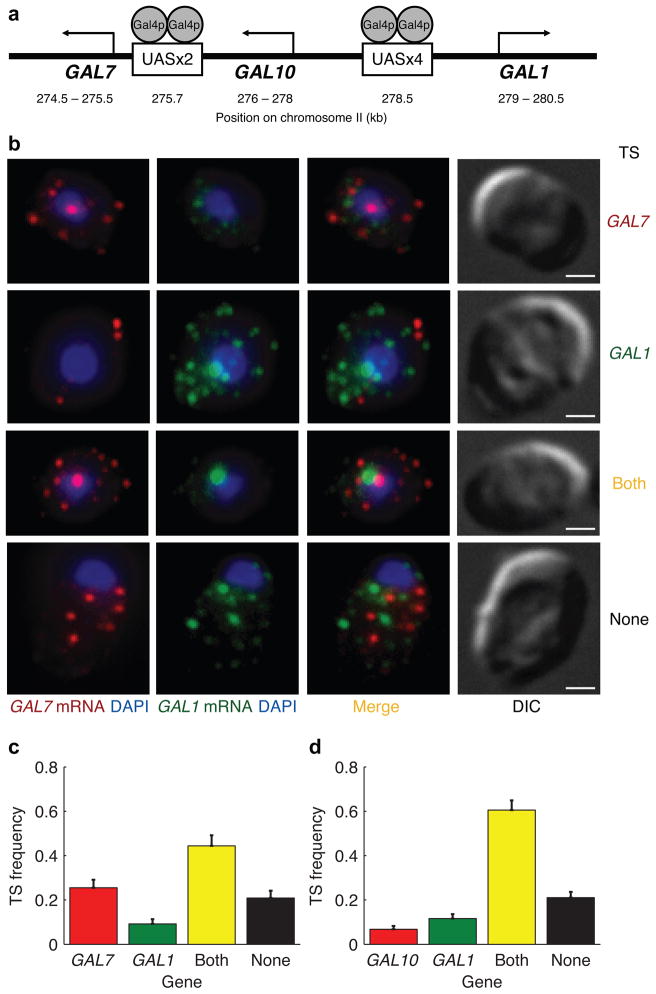
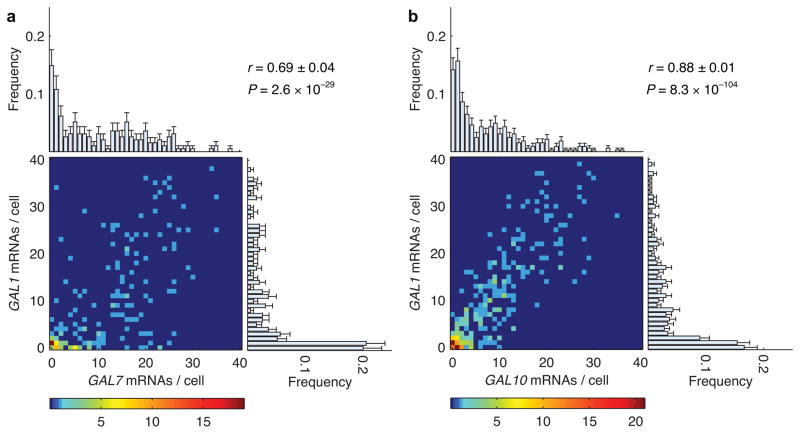
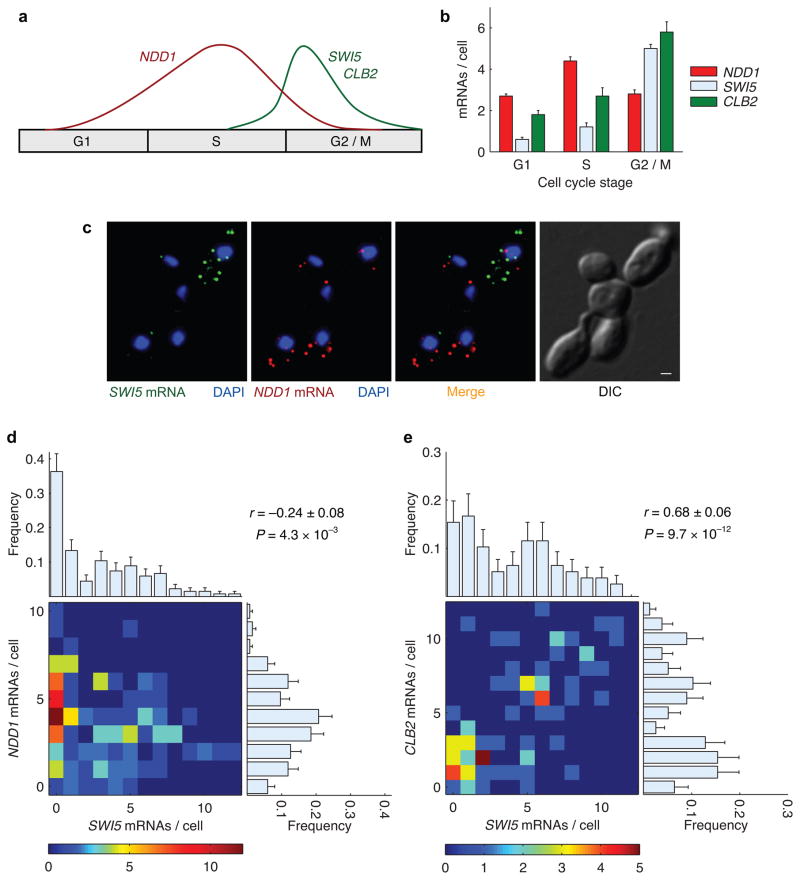
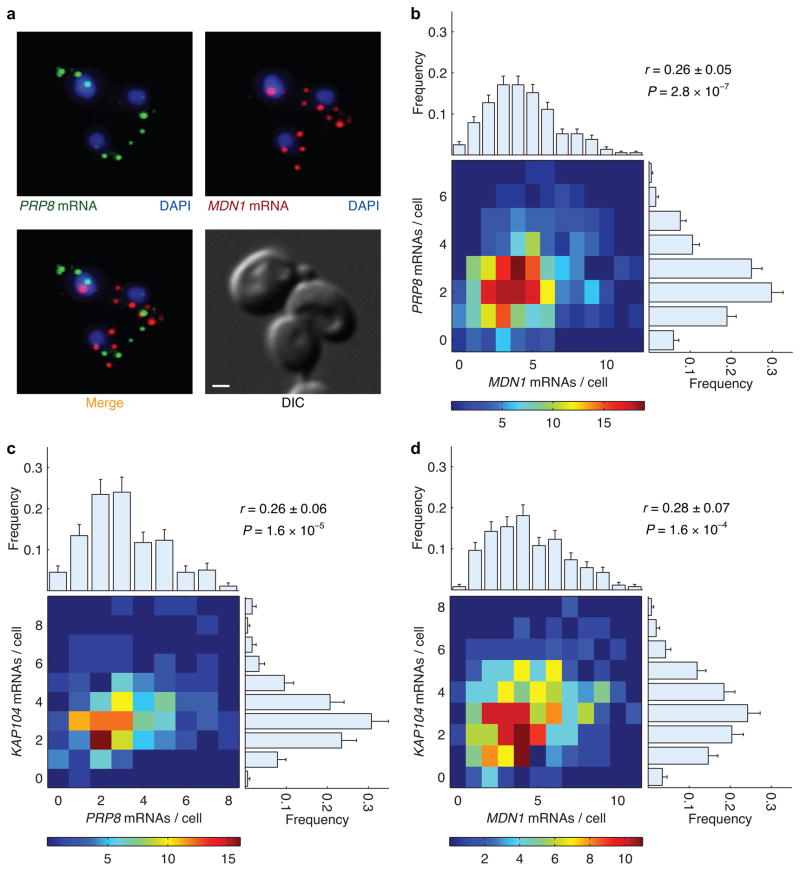
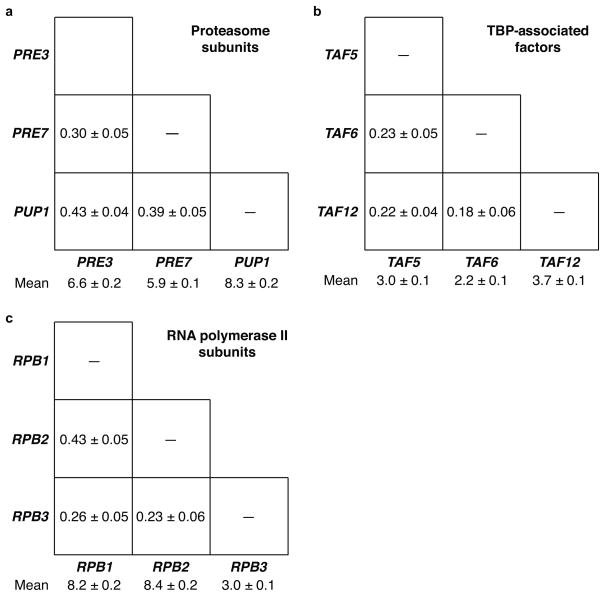
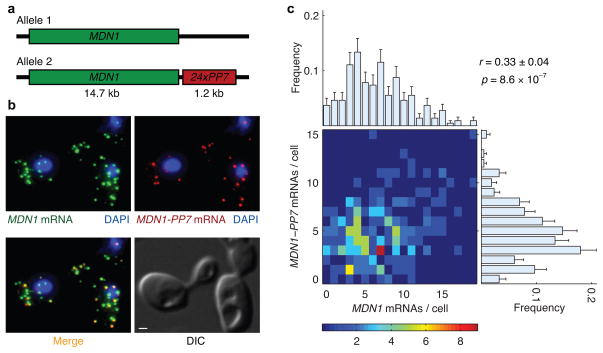
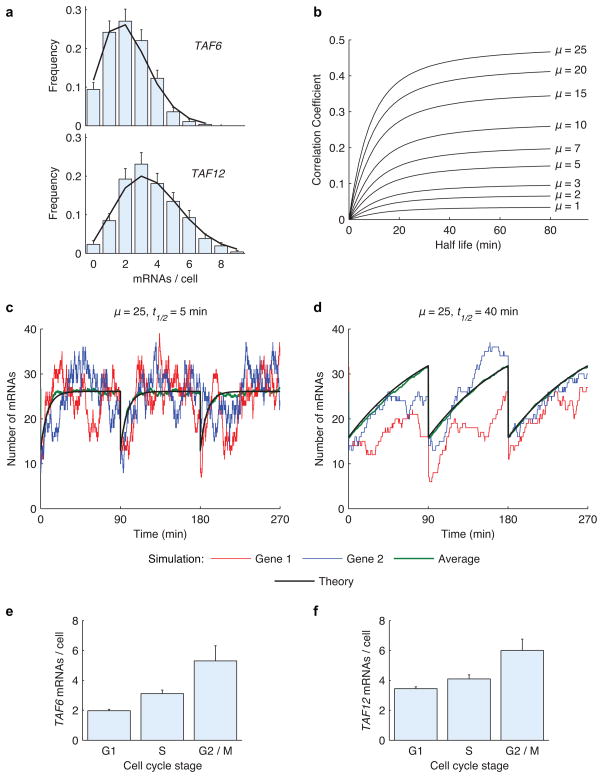
Expression of an individual gene can vary considerably among genetically identical cells because of stochastic fluctuations in transcription. However, proteins comprising essential complexes or pathways have similar abundances and lower variability. It is not known whether coordination in the expression of subunits of essential complexes occurs at the level of transcription, mRNA abundance or protein expression. To directly measure the level of coordination in the expression of genes, we used highly sensitive fluorescence in situ hybridization (FISH) to count individual mRNAs of functionally related and unrelated genes within single Saccharomyces cerevisiae cells. Our results revealed that transcript levels of temporally induced genes are highly correlated in individual cells. In contrast, transcription of constitutive genes encoding essential subunits of complexes is not coordinated because of stochastic fluctuations. The coordination of these functional complexes therefore must occur post-transcriptionally, and likely post-translationally.
Figures
Similar articles
-
Rsp5 is required for the nuclear export of mRNA of HSF1 and MSN2/4 under stress conditions in Saccharomyces cerevisiae.Genes Cells. 2008 Feb;13(2):105-16. doi: 10.1111/j.1365-2443.2007.01154.x. Genes Cells. 2008. PMID: 18233954
-
Measurement and modeling of transcriptional noise in the cell cycle regulatory network.Cell Cycle. 2013 Oct 1;12(19):3203-18. doi: 10.4161/cc.26257. Epub 2013 Sep 4. Cell Cycle. 2013. PMID: 24013422 Free PMC article.
-
Single-RNA counting reveals alternative modes of gene expression in yeast.Nat Struct Mol Biol. 2008 Dec;15(12):1263-71. doi: 10.1038/nsmb.1514. Epub 2008 Nov 16. Nat Struct Mol Biol. 2008. PMID: 19011635 Free PMC article.
-
The role of chromatin structure in regulating stress-induced transcription in Saccharomyces cerevisiae.Biochem Cell Biol. 2006 Aug;84(4):477-89. doi: 10.1139/o06-079. Biochem Cell Biol. 2006. PMID: 16936821 Review.
-
mRNA export and gene expression: the SAGA-TREX-2 connection.Biochim Biophys Acta. 2012 Jun;1819(6):555-65. doi: 10.1016/j.bbagrm.2011.11.011. Epub 2011 Dec 8. Biochim Biophys Acta. 2012. PMID: 22178374 Review.
Cited by
-
What have single-molecule studies taught us about gene expression?Genes Dev. 2016 Aug 15;30(16):1796-810. doi: 10.1101/gad.281725.116. Genes Dev. 2016. PMID: 27601529 Free PMC article. Review.
-
Distribution of Initiation Times Reveals Mechanisms of Transcriptional Regulation in Single Cells.Biophys J. 2018 May 8;114(9):2072-2082. doi: 10.1016/j.bpj.2018.03.031. Biophys J. 2018. PMID: 29742401 Free PMC article.
-
The Low Noise Limit in Gene Expression.PLoS One. 2015 Oct 21;10(10):e0140969. doi: 10.1371/journal.pone.0140969. eCollection 2015. PLoS One. 2015. PMID: 26488303 Free PMC article.
-
Deciphering Transcriptional Dynamics In Vivo by Counting Nascent RNA Molecules.PLoS Comput Biol. 2015 Nov 6;11(11):e1004345. doi: 10.1371/journal.pcbi.1004345. eCollection 2015 Nov. PLoS Comput Biol. 2015. PMID: 26544860 Free PMC article.
-
Single cell transcriptomics: methods and applications.Front Oncol. 2015 Mar 10;5:53. doi: 10.3389/fonc.2015.00053. eCollection 2015. Front Oncol. 2015. PMID: 25806353 Free PMC article. Review.
References
-
- Gavin AC, et al. Proteome survey reveals modularity of the yeast cell machinery. Nature. 2006;440:631–6. - PubMed
-
- Krogan NJ, et al. Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature. 2006;440:637–43. - PubMed
-
- Gavin AC, et al. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature. 2002;415:141–7. - PubMed
-
- Holstege FC, et al. Dissecting the regulatory circuitry of a eukaryotic genome. Cell. 1998;95:717–28. - PubMed
-
- Lee TI, et al. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science. 2002;298:799–804. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
