

Exploiting models of molecular evolution to efficiently direct protein engineering
- PMID: 21132281
- PMCID: PMC3183505
- DOI: 10.1007/s00239-010-9415-2
Exploiting models of molecular evolution to efficiently direct protein engineering
Abstract
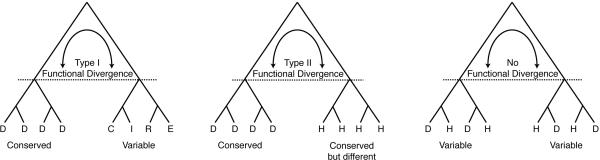
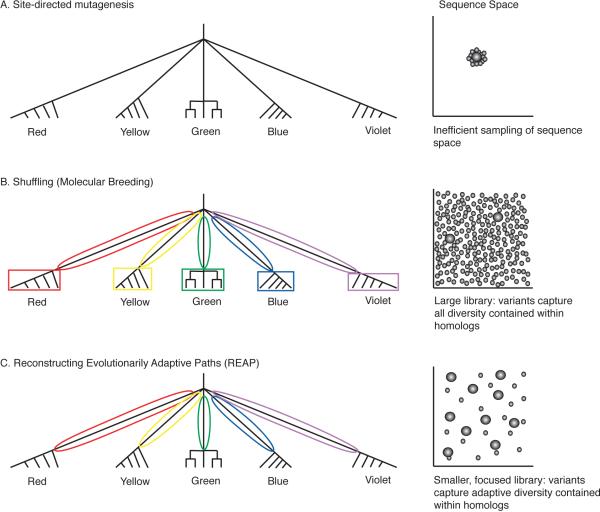
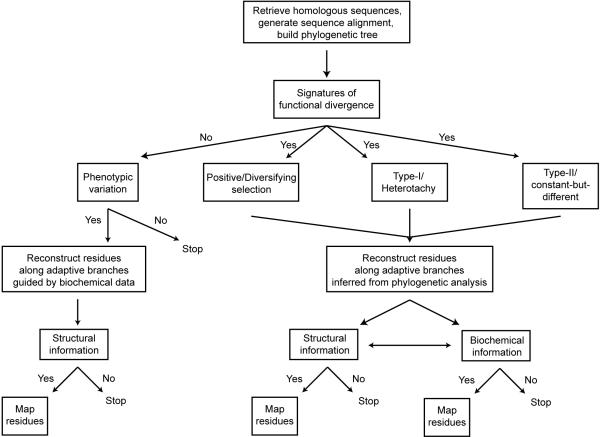
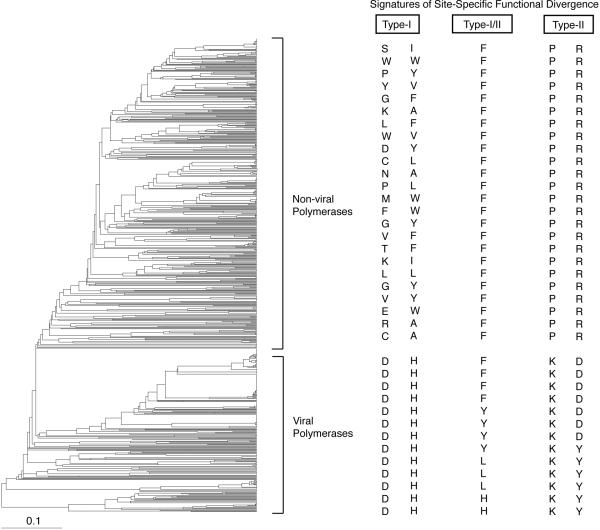
Directed evolution and protein engineering approaches used to generate novel or enhanced biomolecular function often use the evolutionary sequence diversity of protein homologs to rationally guide library design. To fully capture this sequence diversity, however, libraries containing millions of variants are often necessary. Screening libraries of this size is often undesirable due to inaccuracies of high-throughput assays, costs, and time constraints. The ability to effectively cull sequence diversity while still generating the functional diversity within a library thus holds considerable value. This is particularly relevant when high-throughput assays are not amenable to select/screen for certain biomolecular properties. Here, we summarize our recent attempts to develop an evolution-guided approach, Reconstructing Evolutionary Adaptive Paths (REAP), for directed evolution and protein engineering that exploits phylogenetic and sequence analyses to identify amino acid substitutions that are likely to alter or enhance function of a protein. To demonstrate the utility of this technique, we highlight our previous work with DNA polymerases in which a REAP-designed small library was used to identify a DNA polymerase capable of accepting non-standard nucleosides. We anticipate that the REAP approach will be used in the future to facilitate the engineering of biopolymers with expanded functions and will thus have a significant impact on the developing field of 'evolutionary synthetic biology'.
Figures
References
-
- Arnold FH, Georgiou G. Directed Enzyme Evolution: Screening and Selection Methods. Humana Press; Totowa, New Jersey: 2003.
-
- Benner SA, Gaucher EA. Evolution, language and analogy in functional genomics. Trends Genet. 2001;17:414–8. - PubMed
-
- Bielawski JP, Yang Z. A maximum likelihood method for detecting functional divergence at individual codon sites, with application to gene family evolution. J Mol Evol. 2004;59:121–32. - PubMed
-
- Brakmann S. Discovery of superior enzymes by directed molecular evolution. Chembiochem. 2001;2:865–71. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
