

AP-1 regulates cyclin D1 and c-MYC transcription in an AKT-dependent manner in response to mTOR inhibition: role of AIP4/Itch-mediated JUNB degradation
- PMID: 21135252
- PMCID: PMC3105464
- DOI: 10.1158/1541-7786.MCR-10-0105
AP-1 regulates cyclin D1 and c-MYC transcription in an AKT-dependent manner in response to mTOR inhibition: role of AIP4/Itch-mediated JUNB degradation
Abstract
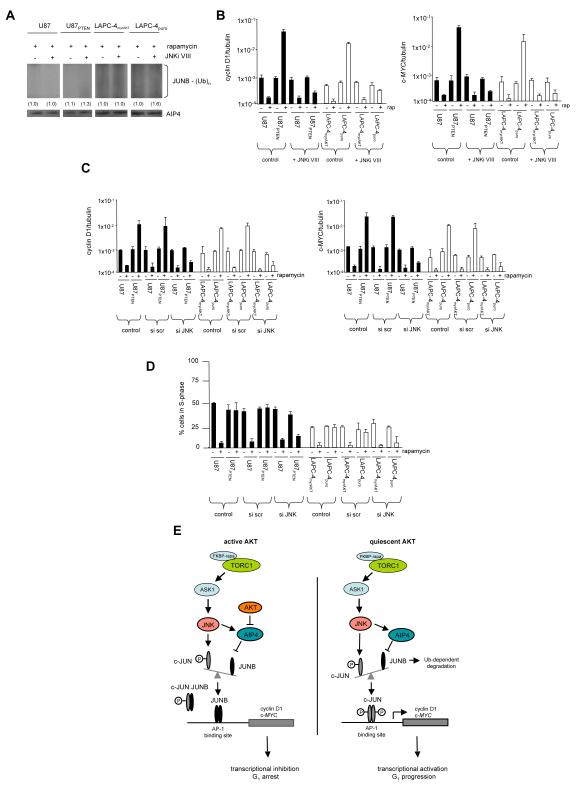
One mechanism by which AKT kinase-dependent hypersensitivity to mammalian target of rapamycin (mTOR) inhibitors is controlled is by the differential expression of cyclin D1 and c-MYC. Regulation of posttranscriptional processes has been demonstrated to be crucial in governing expression of these determinants in response to rapamycin. Our previous data suggested that cyclin D1 and c-MYC expression might additionally be coordinately regulated in an AKT-dependent manner at the level of transcription. Under conditions of relatively quiescent AKT activity, treatment of cells with rapamycin resulted in upregulation of cyclin D1 and c-MYC nascent transcription, whereas in cells containing active AKT, exposure repressed transcription. Promoter analysis identified AKT-dependent rapamycin responsive elements containing AP-1 transactivation sites. Phosphorylated c-JUN binding to these promoters correlated with activation of transcription whereas JUNB occupancy was associated with promoter repression. Forced overexpression of JunB or a conditionally active JunB-ER allele repressed cyclin D1 and c-MYC promoter activity in quiescent AKT-containing cells following rapamycin exposure. AIP4/Itch-dependent JUNB protein degradation was found to be markedly reduced in active AKT-containing cells compared with cells harboring quiescent AKT. Moreover, silencing AIP4/Itch expression or inhibiting JNK mediated AIP4 activity abrogated the rapamycin-induced effects on cyclin D1 and c-MYC promoter activities. Our findings support a role for the AKT-dependent regulation of AIP4/Itch activity in mediating the differential cyclin D1 and c-MYC transcriptional responses to rapamycin.
©2010 AACR.
Figures






References
-
- Guertin DA, Sabatini DM. The pharmacology of mTOR inhibition. Sci Signal. 2009;2:pe24. - PubMed
-
- Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12:9–22. - PubMed
-
- Easton JB, Houghton PJ. mTOR and cancer therapy. Oncogene. 2006;25:6436–46. - PubMed
-
- Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer. 2006;6:729–34. - PubMed
-
- Proud CG. mTORC1 signalling and mRNA translation. Biochem Soc Trans. 2009;37:227–31. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
