

Applications of next generation sequencing in molecular ecology of non-model organisms
- PMID: 21139633
- PMCID: PMC3186121
- DOI: 10.1038/hdy.2010.152
Applications of next generation sequencing in molecular ecology of non-model organisms
Abstract
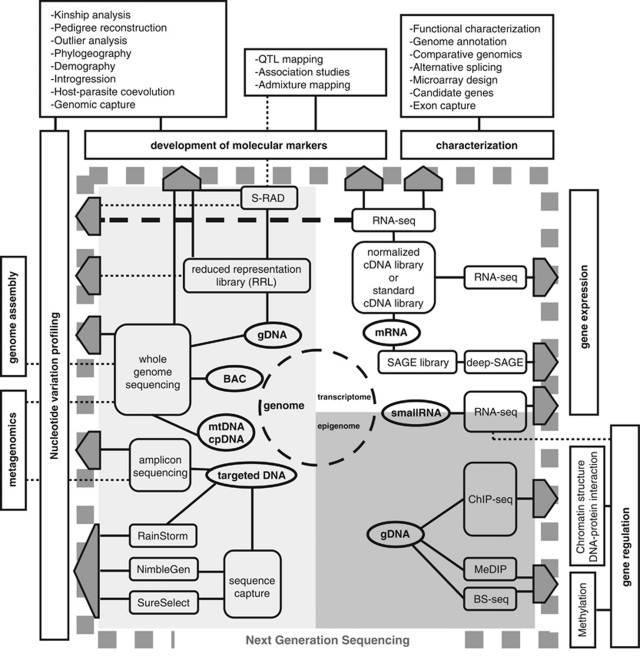
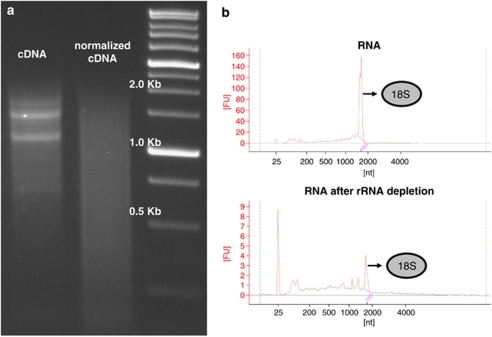
As most biologists are probably aware, technological advances in molecular biology during the last few years have opened up possibilities to rapidly generate large-scale sequencing data from non-model organisms at a reasonable cost. In an era when virtually any study organism can 'go genomic', it is worthwhile to review how this may impact molecular ecology. The first studies to put the next generation sequencing (NGS) to the test in ecologically well-characterized species without previous genome information were published in 2007 and the beginning of 2008. Since then several studies have followed in their footsteps, and a large number are undoubtedly under way. This review focuses on how NGS has been, and can be, applied to ecological, population genetic and conservation genetic studies of non-model species, in which there is no (or very limited) genomic resources. Our aim is to draw attention to the various possibilities that are opening up using the new technologies, but we also highlight some of the pitfalls and drawbacks with these methods. We will try to provide a snapshot of the current state of the art for this rapidly advancing and expanding field of research and give some likely directions for future developments.
Figures
References
-
- Allentoft ME, Schuster SC, Holdaway RN, Hale ML, McLay E, Oskam C, et al. Identification of microsatellites from an extinct moa species using high-throughput (454) sequence data. Biotechniques. 2009;46:195–200. - PubMed
-
- Alvarez LA, Exton DA, Timmis KN, Suggett DJ, McGenity TJ. Characterization of marine isoprene-degrading communities. Environ Microbiol. 2009;11:3280–3291. - PubMed
-
- Andersson AF, Riemann L, Bertilsson S. Pyrosequencing reveals contrasting seasonal dynamics of taxa within Baltic Sea bacterioplankton communities. ISME J. 2010;4:171–181. - PubMed
-
- Babik W, Taberlet P, Ejsmond MJ, Radwan J. New generation sequencers as a tool for genotyping of highly polymorphic multilocus MHC system. Mol Ecol Resour. 2009;9:713–719. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Research Materials
