

Population specificity of the DNAI1 gene mutation spectrum in primary ciliary dyskinesia (PCD)
- PMID: 21143860
- PMCID: PMC3014902
- DOI: 10.1186/1465-9921-11-174
Population specificity of the DNAI1 gene mutation spectrum in primary ciliary dyskinesia (PCD)
Abstract
Background: Mutations in the DNAI1 gene, encoding a component of outer dynein arms of the ciliary apparatus, are the second most important genetic cause of primary ciliary dyskinesia (PCD), the genetically heterogeneous recessive disorder with the prevalence of ~1/20,000. The estimates of the DNAI1 involvement in PCD pathogenesis differ among the reported studies, ranging from 4% to 10%.
Methods: The coding sequence of DNAI1 was screened (SSCP analysis and direct sequencing) in a group of PCD patients (157 families, 185 affected individuals), the first ever studied large cohort of PCD patients of Slavic origin (mostly Polish); multiplex ligation-dependent probe amplification (MLPA) analysis was performed in a subset of ~80 families.
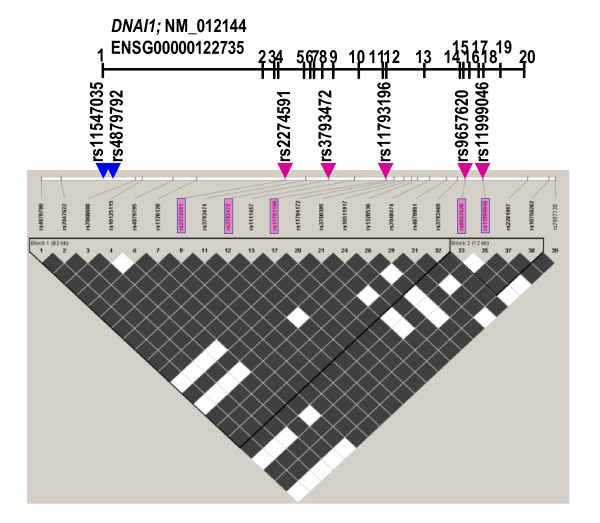
Results: Three previously reported mutations (IVS1+2-3insT, L513P and A538T) and two novel missense substitutions (C388Y and G515S) were identified in 12 families (i.e. ~8% of non-related Polish PCD patients). The structure of background SNP haplotypes indicated common origin of each of the two most frequent mutations, IVS1+2-3insT and A538T. MLPA analysis did not reveal any significant differences between patients and control samples. The Polish cohort was compared with all the previously studied PCD groups (a total of 487 families): IVS1+2-3insT remained the most prevalent pathogenetic change in DNAI1 (54% of the mutations identified worldwide), and the increased global prevalence of A538T (14%) was due to the contribution of the Polish cohort.
Conclusions: The worldwide involvement of DNAI1 mutations in PCD pathogenesis in families not preselected for ODA defects ranges from 7 to 10%; this global estimate as well as the mutation profile differs in specific populations. Analysis of the background SNP haplotypes suggests that the increased frequency of chromosomes carrying A538T mutations in Polish patients may reflects local (Polish or Slavic) founder effect. Results of the MLPA analysis indicate that no large exonic deletions are involved in PCD pathogenesis.
Figures




References
-
- Afzelius BA, Mossberg B. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill, Inc; 1995. Immotile-cilia syndrome (primary ciliary dyskinesia), including Kartagener syndrome; pp. 3943–54.
-
- Krawczyński MR, Witt M. PCD and RP: X-linked inheritance of both disorders? Pediatr Pulmonol. 2004;38:88–9. - PubMed
-
- Krawczyński MR, Dmeńska H, Witt M. Apparent X-linked primary ciliary dyskinesia associated with retinitis pigmentosa and a hearing loss. J Appl Genet. 2004;45:107–10. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
