

Risk of colorectal and endometrial cancers in EPCAM deletion-positive Lynch syndrome: a cohort study
- PMID: 21145788
- PMCID: PMC3670774
- DOI: 10.1016/S1470-2045(10)70265-5
Risk of colorectal and endometrial cancers in EPCAM deletion-positive Lynch syndrome: a cohort study
Abstract
Background: Lynch syndrome is caused by germline mutations in MSH2, MLH1, MSH6, and PMS2 mismatch-repair genes and leads to a high risk of colorectal and endometrial cancer. We previously showed that constitutional 3' end deletions of EPCAM can cause Lynch syndrome through epigenetic silencing of MSH2 in EPCAM-expressing tissues, resulting in tissue-specific MSH2 deficiency. We aim to establish the risk of cancer associated with such EPCAM deletions.
Methods: We obtained clinical data for 194 carriers of a 3' end EPCAM deletion from 41 families known to us at the Radboud University Nijmegen Medical Centre, Nijmegen, Netherlands and compared cancer risk with data from a previously described cohort of 473 carriers from 91 families with mutations in MLH1, MSH2, MSH6, or a combined EPCAM-MSH2 deletion.
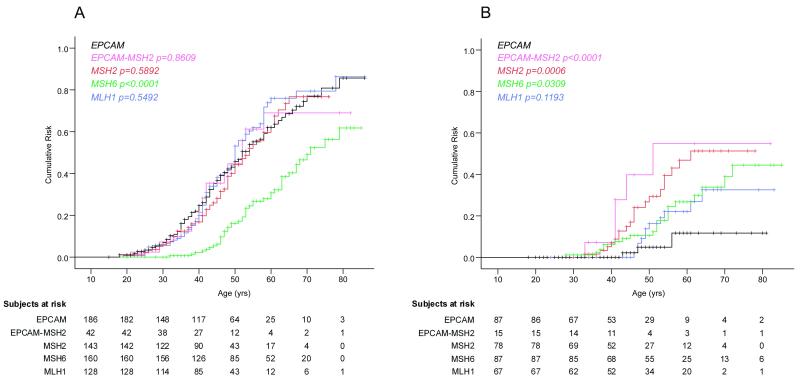
Findings: 93 of the 194 EPCAM deletion carriers were diagnosed with colorectal cancer; three of the 92 women with EPCAM deletions were diagnosed with endometrial cancer. Carriers of an EPCAM deletion had a 75% (95% CI 65-85) cumulative risk of colorectal cancer before the age of 70 years (mean age at diagnosis 43 years [SD 12]), which did not differ significantly from that of carriers of combined EPCAM-MSH2 deletion (69% [95% CI 47-91], p=0·8609) or mutations in MSH2 (77% [64-90], p=0·5892) or MLH1 (79% [68-90], p=0·5492), but was higher than noted for carriers of MSH6 mutation (50% [38-62], p<0·0001). By contrast, women with EPCAM deletions had a 12% [0-27] cumulative risk of endometrial cancer, which was lower than was that noted for carriers of a combined EPCAM-MSH2 deletion (55% [20-90], p<0·0001) or of a mutation in MSH2 (51% [33-69], p=0·0006) or MSH6 (34% [20-48], p=0·0309), but did not differ significantly from that noted for MLH1 (33% [15-51], p=0·1193) mutation carriers. This risk seems to be restricted to deletions that extend close to the MSH2 gene promoter. Of 194 carriers of an EPCAM deletion, three had duodenal cancer and four had pancreatic cancer.
Interpretation: EPCAM deletion carriers have a high risk of colorectal cancer; only those with deletions extending close to the MSH2 promoter have an increased risk of endometrial cancer. These results underscore the effect of mosaic MSH2 deficiency, leading to variable cancer risks, and could form the basis of an optimised protocol for the recognition and targeted prevention of cancer in EPCAM deletion carriers.
Copyright © 2011 Elsevier Ltd. All rights reserved.
Figures

Comment in
-
EPCAM deletions, Lynch syndrome, and cancer risk.Lancet Oncol. 2011 Jan;12(1):5-6. doi: 10.1016/S1470-2045(10)70291-6. Lancet Oncol. 2011. PMID: 21195320 No abstract available.
References
-
- Koornstra JJ, Mourits MJ, Sijmons RH, Leliveld AM, Hollema H, Kleibeuker JH. Management of extracolonic tumours in patients with Lynch syndrome. Lancet Oncol. 2009;10:400–8. - PubMed
-
- Hendriks YM, Wagner A, Morreau H, Menko F, Stormorken A, Quehenberger F, et al. Cancer risk in hereditary nonpolyposis colorectal cancer due to MSH6 mutations: impact on counseling and surveillance. Gastroenterology. 2004;127:17–25. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
