

Endoplasmic reticulum stress in liver disease
- PMID: 21145844
- PMCID: PMC3375108
- DOI: 10.1016/j.jhep.2010.11.005
Endoplasmic reticulum stress in liver disease
Abstract
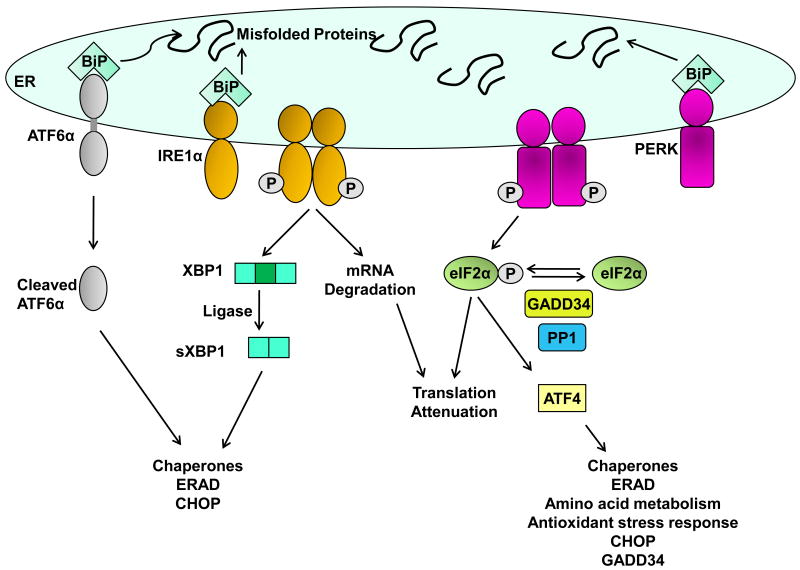
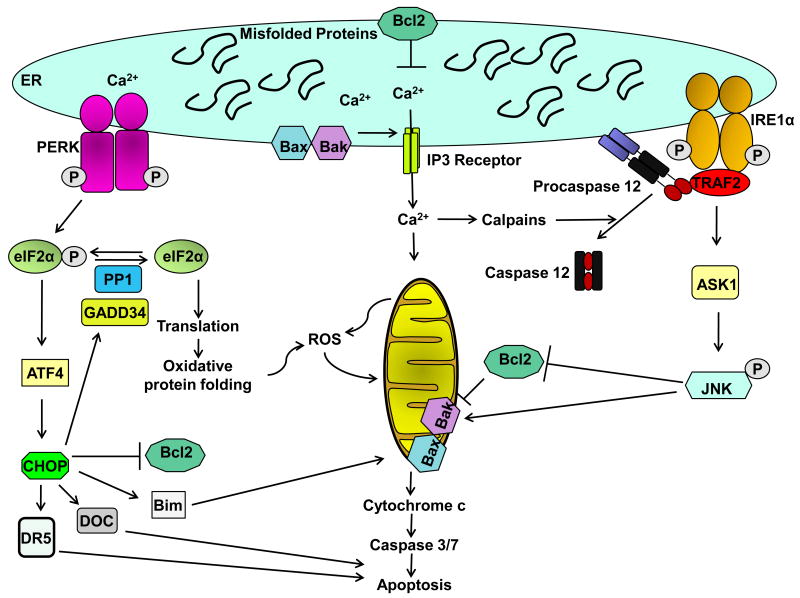
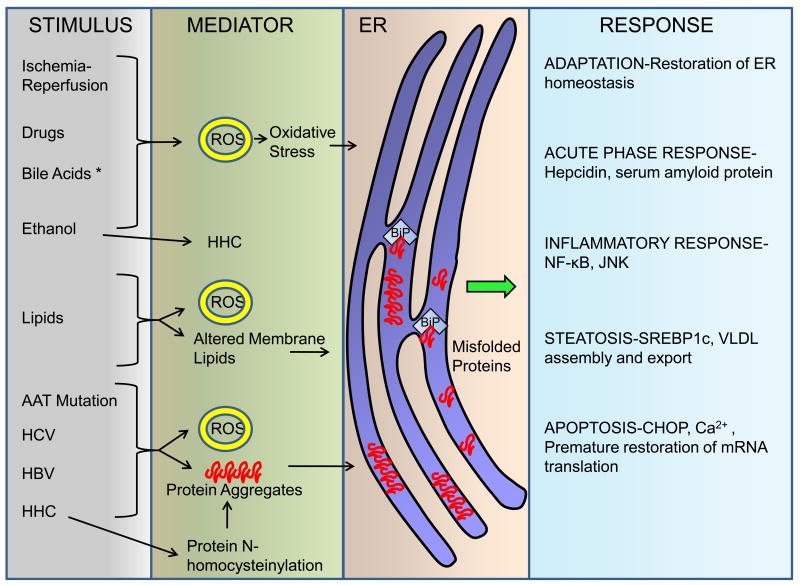
The unfolded protein response (UPR) is activated upon the accumulation of misfolded proteins in the endoplasmic reticulum (ER) that are sensed by the binding immunoglobulin protein (BiP)/glucose-regulated protein 78 (GRP78). The accumulation of unfolded proteins sequesters BiP so it dissociates from three ER-transmembrane transducers leading to their activation. These transducers are inositol requiring (IRE) 1α, PKR-like ER kinase (PERK), and activating transcription factor (ATF) 6α. PERK phosphorylates eukaryotic initiation factor 2 alpha (eIF2α) resulting in global mRNA translation attenuation, and concurrently selectively increases the translation of several mRNAs, including the transcription factor ATF4, and its downstream target CHOP. IRE1α has kinase and endoribonuclease (RNase) activities. IRE1α autophosphorylation activates the RNase activity to splice XBP1 mRNA, to produce the active transcription factor sXBP1. IRE1α activation also recruits and activates the stress kinase JNK. ATF6α transits to the Golgi compartment where it is cleaved by intramembrane proteolysis to generate a soluble active transcription factor. These UPR pathways act in concert to increase ER content, expand the ER protein folding capacity, degrade misfolded proteins, and reduce the load of new proteins entering the ER. All of these are geared toward adaptation to resolve the protein folding defect. Faced with persistent ER stress, adaptation starts to fail and apoptosis occurs, possibly mediated through calcium perturbations, reactive oxygen species, and the proapoptotic transcription factor CHOP. The UPR is activated in several liver diseases; including obesity associated fatty liver disease, viral hepatitis, and alcohol-induced liver injury, all of which are associated with steatosis, raising the possibility that ER stress-dependent alteration in lipid homeostasis is the mechanism that underlies the steatosis. Hepatocyte apoptosis is a pathogenic event in several liver diseases, and may be linked to unresolved ER stress. If this is true, restoration of ER homeostasis prior to ER stress-induced cell death may provide a therapeutic rationale in these diseases. Herein we discuss each branch of the UPR and how they may impact hepatocyte function in different pathologic states.
Copyright © 2010 European Association for the Study of the Liver. Published by Elsevier B.V. All rights reserved.
Conflict of interest statement
Conflict of Interest: The authors declare that they have no conflict of interest with respect to the information presented in this manuscript.
Figures
References
-
- Asselah T, Bieche I, Mansouri A, Laurendeau I, Cazals-Hatem D, Feldmann G, et al. In vivo hepatic endoplasmic reticulum stress in patients with chronic hepatitis C. J Pathol. 2010;221(3):264–274. - PubMed
-
- Awe K, Lambert C, Prange R. Mammalian BiP controls posttranslational ER translocation of the hepatitis B virus large envelope protein. FEBS Lett. 2008;582(21-22):3179–3184. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 DK088227/DK/NIDDK NIH HHS/United States
- P01 HL057346/HL/NHLBI NIH HHS/United States
- DK042394/DK/NIDDK NIH HHS/United States
- T32 DK007198/DK/NIDDK NIH HHS/United States
- HL052173/HL/NHLBI NIH HHS/United States
- R37 DK042394/DK/NIDDK NIH HHS/United States
- P30 DK084567/DK/NIDDK NIH HHS/United States
- HL057346/HL/NHLBI NIH HHS/United States
- T32 DK07198/DK/NIDDK NIH HHS/United States
- R03 MH089782/MH/NIMH NIH HHS/United States
- R01 DK042394/DK/NIDDK NIH HHS/United States
- R01 HL052173/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
