

COMODO: an adaptive coclustering strategy to identify conserved coexpression modules between organisms
- PMID: 21149270
- PMCID: PMC3074154
- DOI: 10.1093/nar/gkq1275
COMODO: an adaptive coclustering strategy to identify conserved coexpression modules between organisms
Abstract
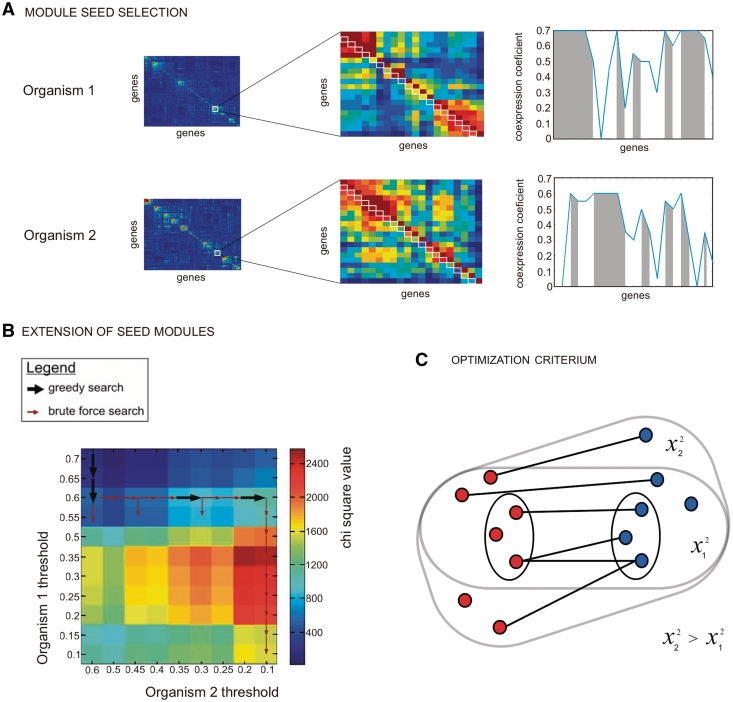
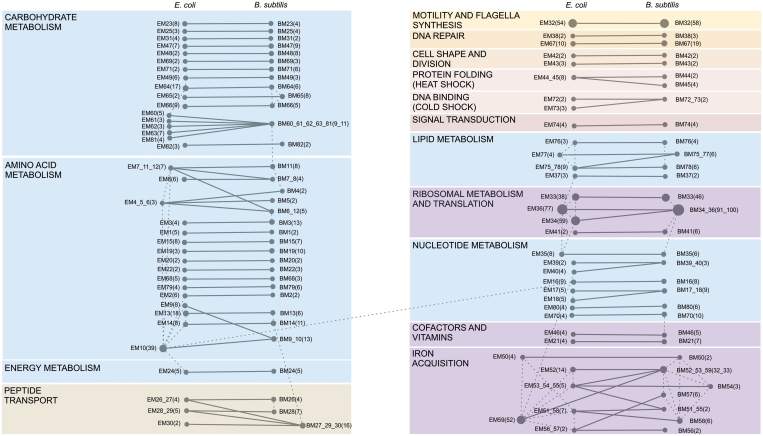
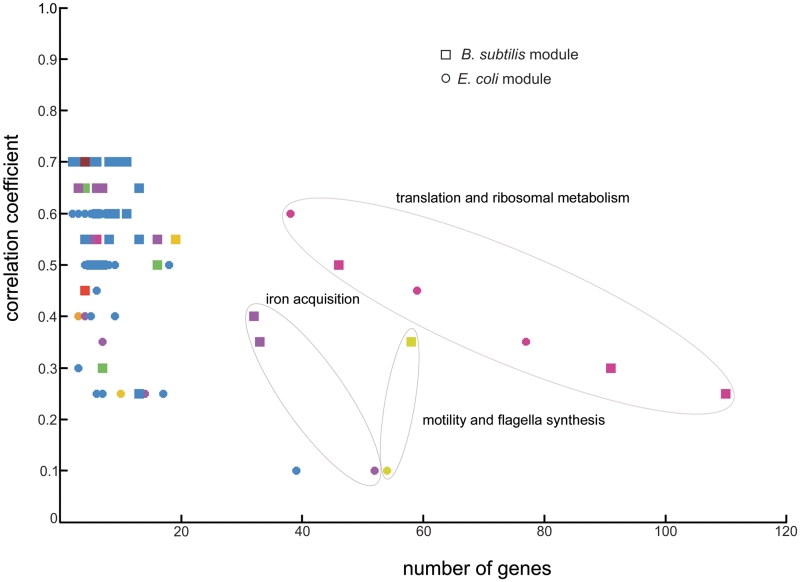
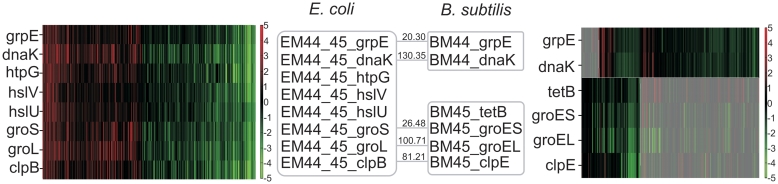
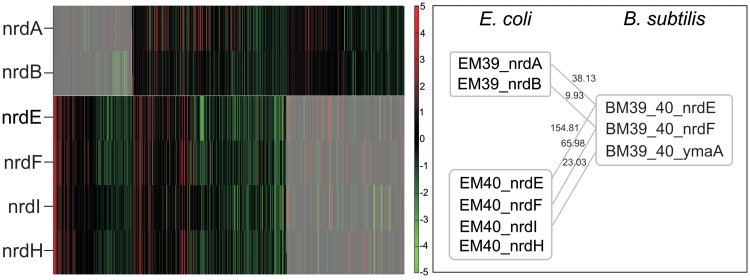
Increasingly large-scale expression compendia for different species are becoming available. By exploiting the modularity of the coexpression network, these compendia can be used to identify biological processes for which the expression behavior is conserved over different species. However, comparing module networks across species is not trivial. The definition of a biologically meaningful module is not a fixed one and changing the distance threshold that defines the degree of coexpression gives rise to different modules. As a result when comparing modules across species, many different partially overlapping conserved module pairs across species exist and deciding which pair is most relevant is hard. Therefore, we developed a method referred to as conserved modules across organisms (COMODO) that uses an objective selection criterium to identify conserved expression modules between two species. The method uses as input microarray data and a gene homology map and provides as output pairs of conserved modules and searches for the pair of modules for which the number of sharing homologs is statistically most significant relative to the size of the linked modules. To demonstrate its principle, we applied COMODO to study coexpression conservation between the two well-studied bacteria Escherichia coli and Bacillus subtilis. COMODO is available at: http://homes.esat.kuleuven.be/∼kmarchal/Supplementary_Information_Zarrineh_2010/comodo/index.html.
Figures

References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
