

LSH and G9a/GLP complex are required for developmentally programmed DNA methylation
- PMID: 21149390
- PMCID: PMC3012929
- DOI: 10.1101/gr.108498.110
LSH and G9a/GLP complex are required for developmentally programmed DNA methylation
Abstract
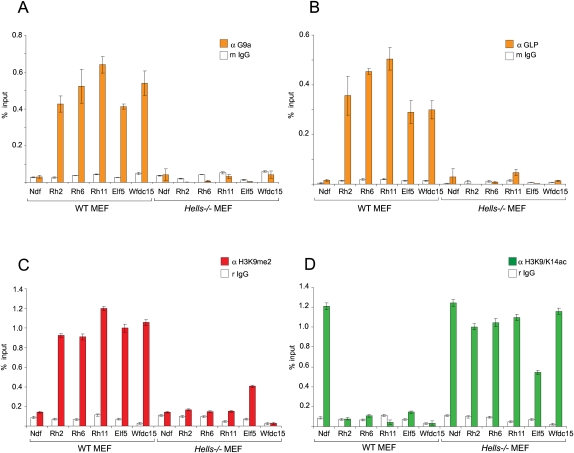
LSH, a member of the SNF2 family of chromatin remodeling ATPases encoded by the Hells gene, is essential for normal levels of DNA methylation in the mammalian genome. While the role of LSH in the methylation of repetitive DNA sequences is well characterized, its contribution to the regulation of DNA methylation and the expression of protein-coding genes has not been studied in detail. In this report we investigate genome-wide patterns of DNA methylation at gene promoters in Hells(-/-) mouse embryonic fibroblasts (MEFs). We find that in the absence of LSH, DNA methylation is lost or significantly reduced at ∼20% of all normally methylated promoter sequences. As a consequence, a large number of genes are misexpressed in Hells(-/-) MEFs. Comparison of Hells(-/-) MEFs with wild-type MEFs and embryonic stem (ES) cells suggests that LSH is important for de novo DNA methylation events that accompany the establishment and differentiation of embryonic lineage cells. We further show that the generation of normal DNA methylation patterns and stable gene silencing at specific promoters require cooperation between LSH and the G9a/GLP complex of histone methylases. At such loci, G9a recruitment is compromised when LSH is absent or greatly reduced. Taken together, our data suggest a mechanism whereby LSH promotes binding of DNA methyltransferases and the G9a/GLP complex to specific loci and facilitates developmentally programmed DNA methylation and stable gene silencing during lineage commitment and differentiation.
Figures






References
-
- Becker PB, Horz W 2002. ATP-dependent nucleosome remodeling. Annu Rev Biochem 71: 247–273 - PubMed
-
- Bird A 2002. DNA methylation patterns and epigenetic memory. Genes Dev 16: 6–21 - PubMed
-
- Brzeski J, Jerzmanowski A 2003. Deficient in DNA methylation 1 (DDM1) defines a novel family of chromatin-remodeling factors. J Biol Chem 278: 823–828 - PubMed
-
- Cross SH, Charlton JA, Nan X, Bird AP 1994. Purification of CpG islands using a methylated DNA binding column. Nat Genet 6: 236–244 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials