

Formin follows function: a muscle-specific isoform of FHOD3 is regulated by CK2 phosphorylation and promotes myofibril maintenance
- PMID: 21149568
- PMCID: PMC3002041
- DOI: 10.1083/jcb.201005060
Formin follows function: a muscle-specific isoform of FHOD3 is regulated by CK2 phosphorylation and promotes myofibril maintenance
Abstract
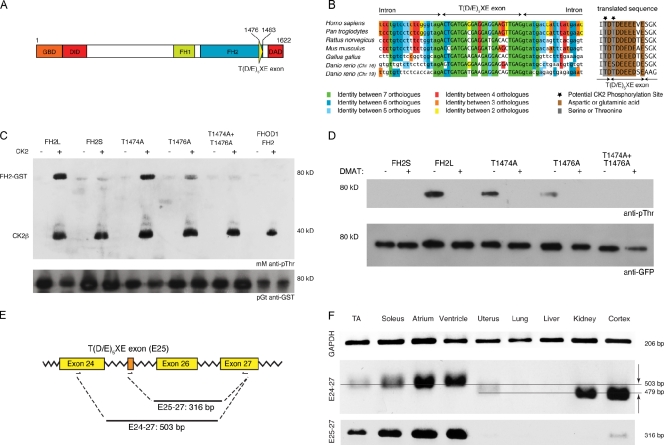
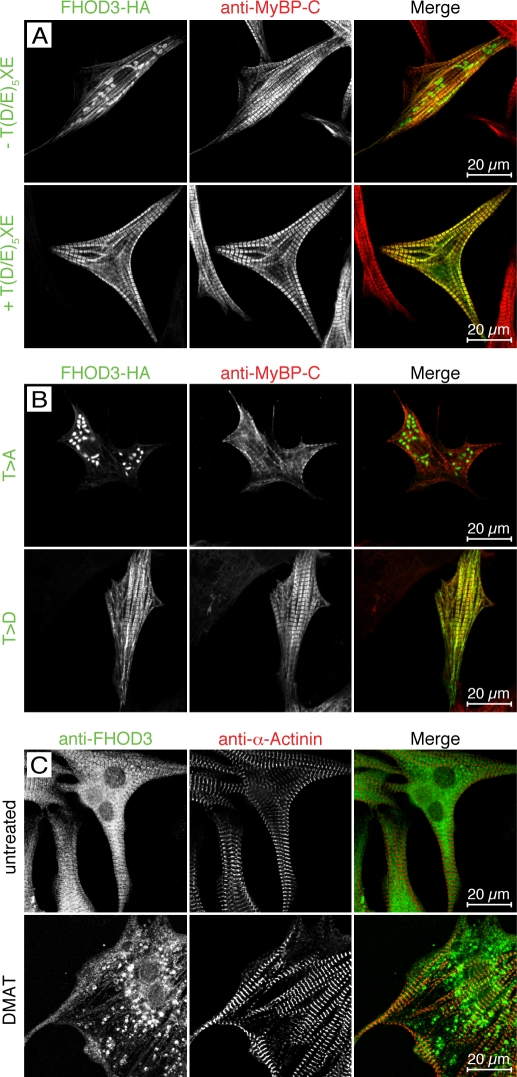
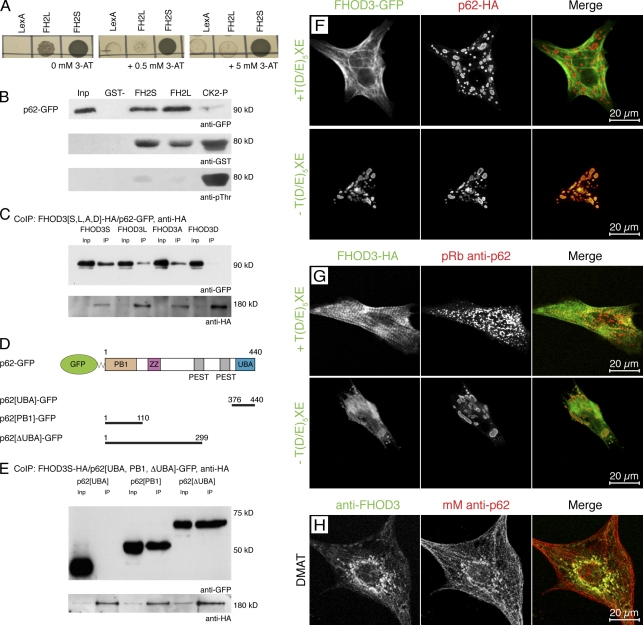
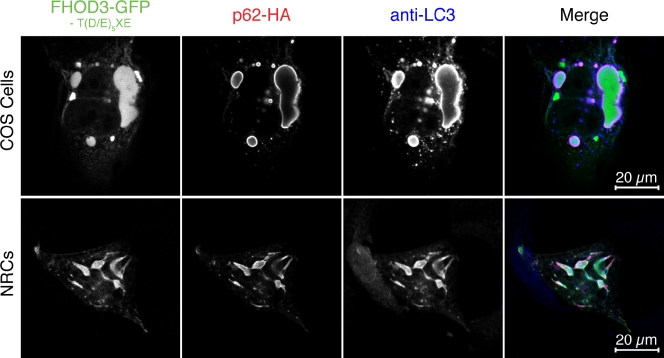
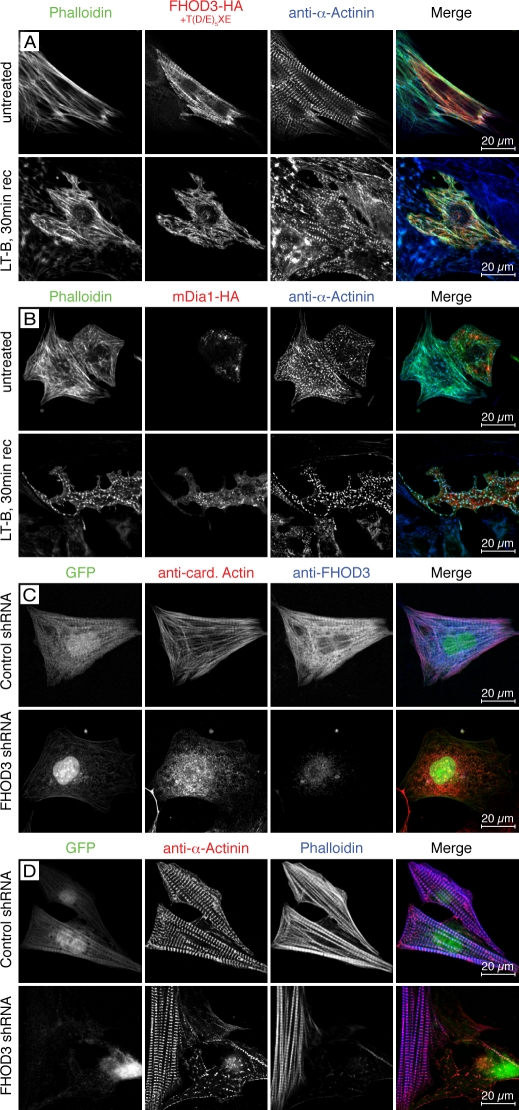
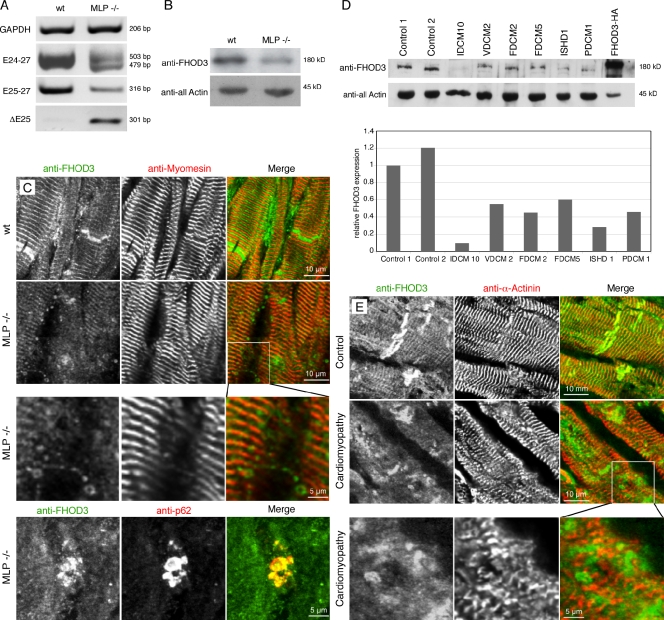
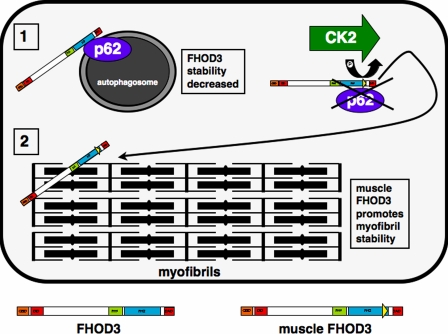
Members of the formin family are important for actin filament nucleation and elongation. We have identified a novel striated muscle-specific splice variant of the formin FHOD3 that introduces a casein kinase 2 (CK2) phosphorylation site. The specific targeting of muscle FHOD3 to the myofibrils in cardiomyocytes is abolished in phosphomutants or by the inhibition of CK2. Phosphorylation of muscle FHOD3 also prevents its interaction with p62/sequestosome 1 and its recruitment to autophagosomes. Furthermore, we show that muscle FHOD3 efficiently promotes the polymerization of actin filaments in cardiomyocytes and that the down-regulation of its expression severely affects myofibril integrity. In murine and human cardiomyopathy, we observe reduced FHOD3 expression with a concomitant isoform switch and change of subcellular targeting. Collectively, our data suggest that a muscle-specific isoform of FHOD3 is required for the maintenance of the contractile structures in heart muscle and that its function is regulated by posttranslational modification.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
