

De novo transcriptome sequencing in Anopheles funestus using Illumina RNA-seq technology
- PMID: 21151993
- PMCID: PMC2996306
- DOI: 10.1371/journal.pone.0014202
De novo transcriptome sequencing in Anopheles funestus using Illumina RNA-seq technology
Abstract
Background: Anopheles funestus is one of the primary vectors of human malaria, which causes a million deaths each year in sub-Saharan Africa. Few scientific resources are available to facilitate studies of this mosquito species and relatively little is known about its basic biology and evolution, making development and implementation of novel disease control efforts more difficult. The An. funestus genome has not been sequenced, so in order to facilitate genome-scale experimental biology, we have sequenced the adult female transcriptome of An. funestus from a newly founded colony in Burkina Faso, West Africa, using the Illumina GAIIx next generation sequencing platform.
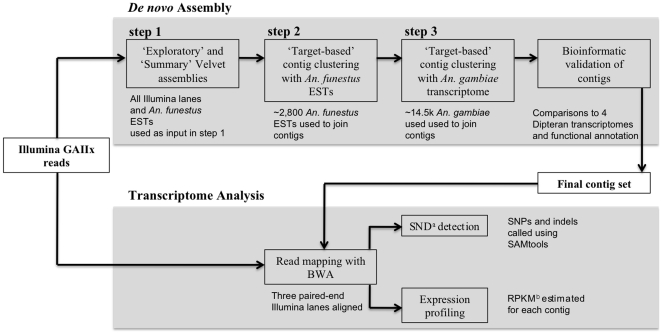
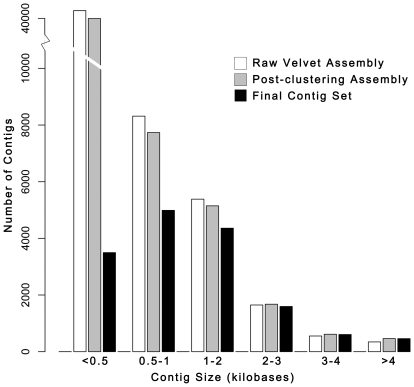
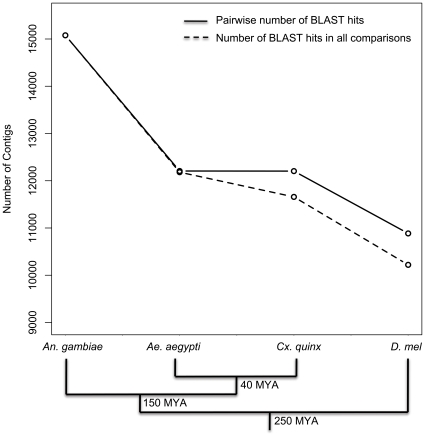
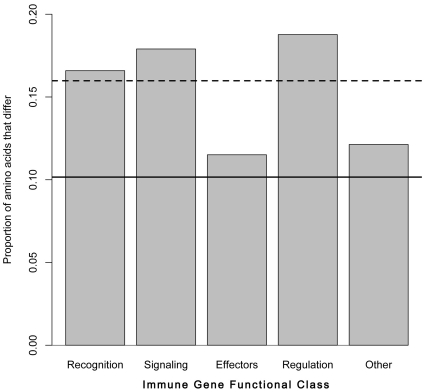
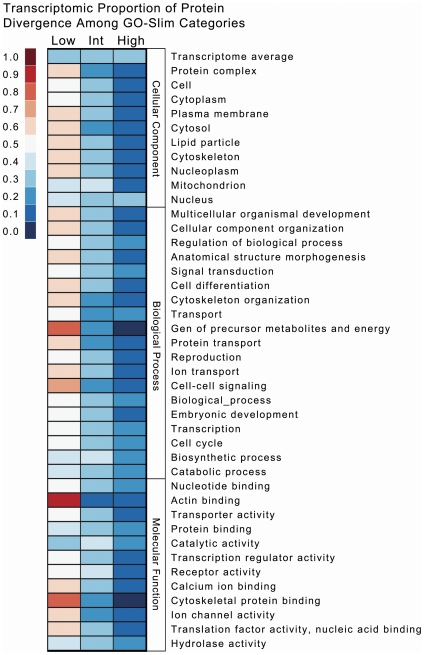
Methodology/principal findings: We assembled short Illumina reads de novo using a novel approach involving iterative de novo assemblies and "target-based" contig clustering. We then selected a conservative set of 15,527 contigs through comparisons to four Dipteran transcriptomes as well as multiple functional and conserved protein domain databases. Comparison to the Anopheles gambiae immune system identified 339 contigs as putative immune genes, thus identifying a large portion of the immune system that can form the basis for subsequent studies of this important malaria vector. We identified 5,434 1:1 orthologues between An. funestus and An. gambiae and found that among these 1:1 orthologues, the protein sequence of those with putative immune function were significantly more diverged than the transcriptome as a whole. Short read alignments to the contig set revealed almost 367,000 genetic polymorphisms segregating in the An. funestus colony and demonstrated the utility of the assembled transcriptome for use in RNA-seq based measurements of gene expression.
Conclusions/significance: We developed a pipeline that makes de novo transcriptome sequencing possible in virtually any organism at a very reasonable cost ($6,300 in sequencing costs in our case). We anticipate that our approach could be used to develop genomic resources in a diversity of systems for which full genome sequence is currently unavailable. Our An. funestus contig set and analytical results provide a valuable resource for future studies in this non-model, but epidemiologically critical, vector insect.
Conflict of interest statement
Figures
References
-
- WHO/UNICEF World Malaria Report. Geneva: World Health Organization; 2009.
-
- Anopheles Genomes Cluster Committee. Genome analysis of vectorial capacity in major Anopheles vectors of malaria parasites. 2008. VectorBase.org.
-
- Hudson HE. Sequencing breakthroughs for genomic ecology and evolutionary biology. Molecular Ecology Resources. 2008;8:3–17. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
