

Identifying a high fraction of the human genome to be under selective constraint using GERP++
- PMID: 21152010
- PMCID: PMC2996323
- DOI: 10.1371/journal.pcbi.1001025
Identifying a high fraction of the human genome to be under selective constraint using GERP++
Abstract
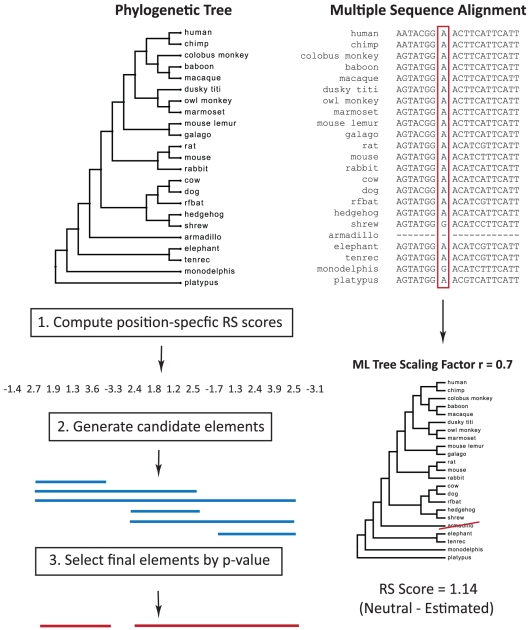
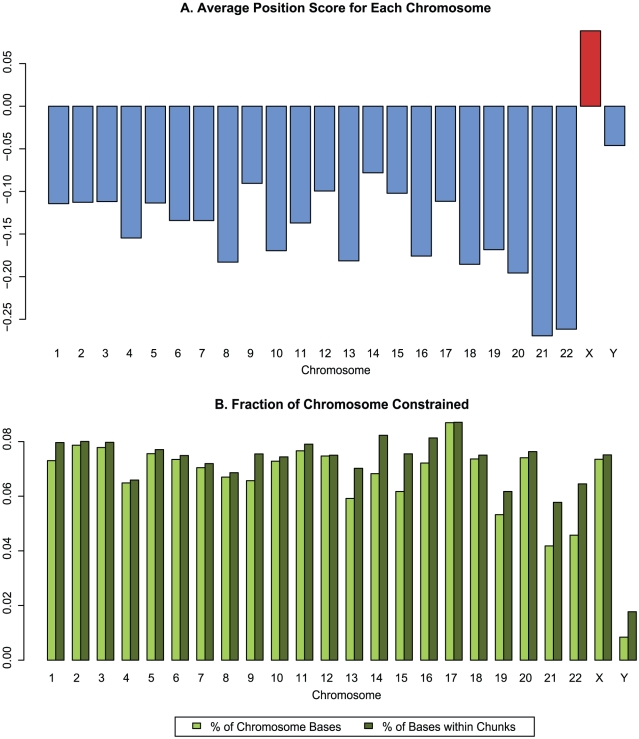
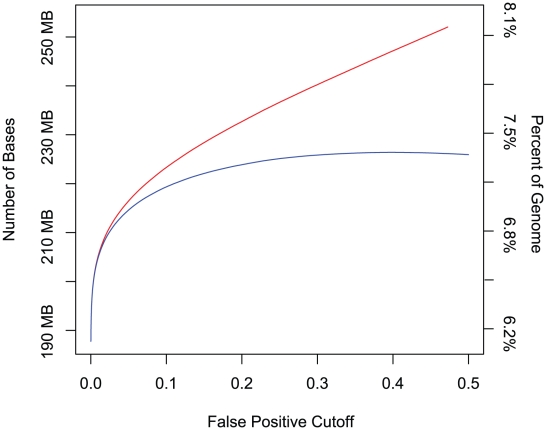
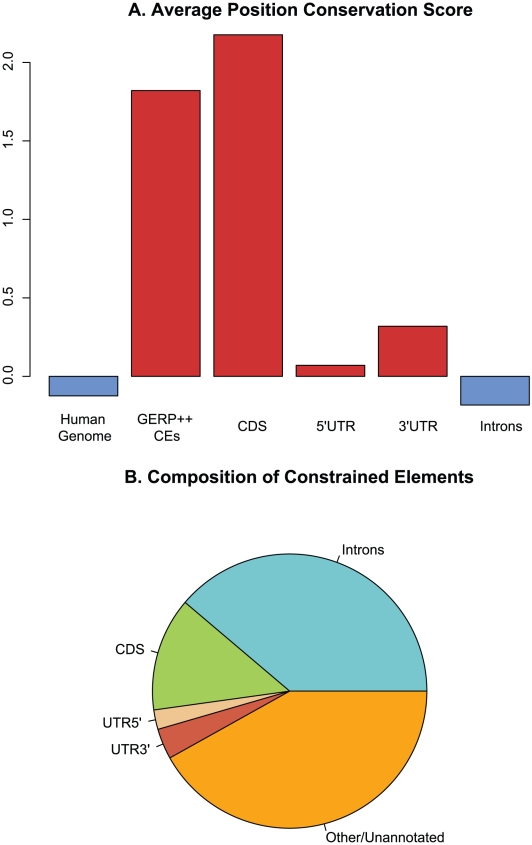
Computational efforts to identify functional elements within genomes leverage comparative sequence information by looking for regions that exhibit evidence of selective constraint. One way of detecting constrained elements is to follow a bottom-up approach by computing constraint scores for individual positions of a multiple alignment and then defining constrained elements as segments of contiguous, highly scoring nucleotide positions. Here we present GERP++, a new tool that uses maximum likelihood evolutionary rate estimation for position-specific scoring and, in contrast to previous bottom-up methods, a novel dynamic programming approach to subsequently define constrained elements. GERP++ evaluates a richer set of candidate element breakpoints and ranks them based on statistical significance, eliminating the need for biased heuristic extension techniques. Using GERP++ we identify over 1.3 million constrained elements spanning over 7% of the human genome. We predict a higher fraction than earlier estimates largely due to the annotation of longer constrained elements, which improves one to one correspondence between predicted elements with known functional sequences. GERP++ is an efficient and effective tool to provide both nucleotide- and element-level constraint scores within deep multiple sequence alignments.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
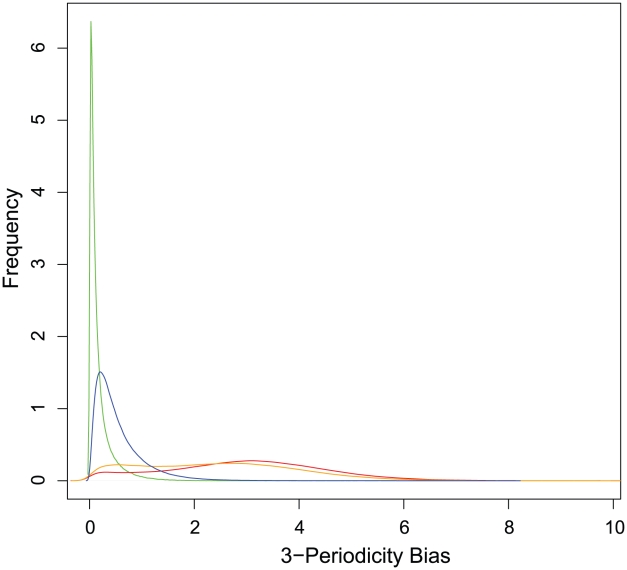
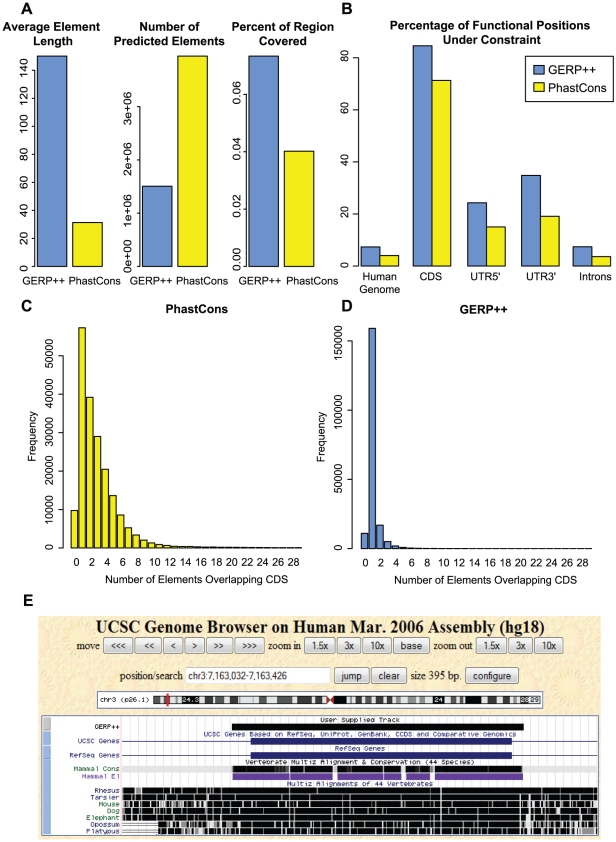
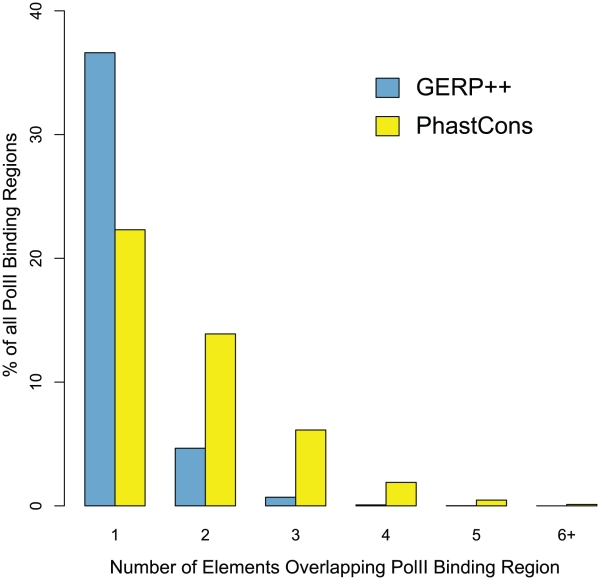
References
-
- The ENCODE Project Consortium. The ENCODE (ENCyclopedia Of DNA Elements) Project. Science. 2004;306:636–640. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
