

Efficient double fragmentation ChIP-seq provides nucleotide resolution protein-DNA binding profiles
- PMID: 21152096
- PMCID: PMC2994895
- DOI: 10.1371/journal.pone.0015092
Efficient double fragmentation ChIP-seq provides nucleotide resolution protein-DNA binding profiles
Abstract
Immunoprecipitated crosslinked protein-DNA fragments typically range in size from several hundred to several thousand base pairs, with a significant part of chromatin being much longer than the optimal length for next-generation sequencing (NGS) procedures. Because these larger fragments may be non-random and represent relevant biology that may otherwise be missed, but also because they represent a significant fraction of the immunoprecipitated material, we designed a double-fragmentation ChIP-seq procedure. After conventional crosslinking and immunoprecipitation, chromatin is de-crosslinked and sheared a second time to concentrate fragments in the optimal size range for NGS. Besides the benefits of increased chromatin yields, the procedure also eliminates a laborious size-selection step. We show that the double-fragmentation ChIP-seq approach allows for the generation of biologically relevant genome-wide protein-DNA binding profiles from sub-nanogram amounts of TCF7L2/TCF4, TBP and H3K4me3 immunoprecipitated material. Although optimized for the AB/SOLiD platform, the same approach may be applied to other platforms.
Conflict of interest statement
Figures
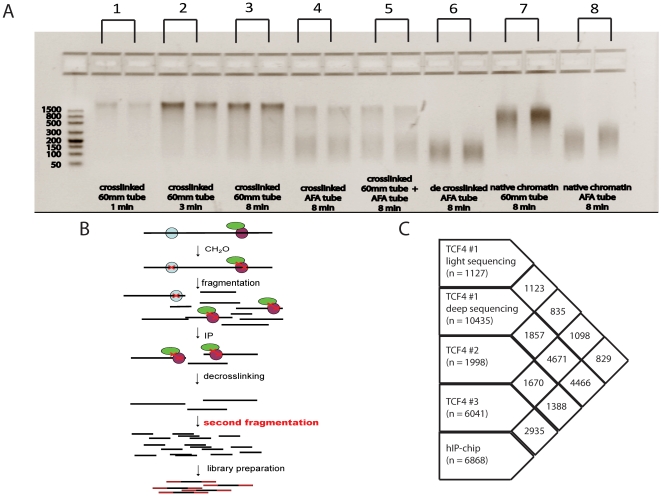
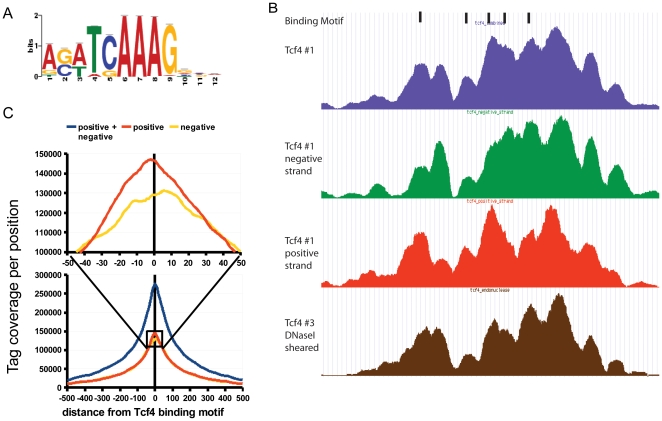
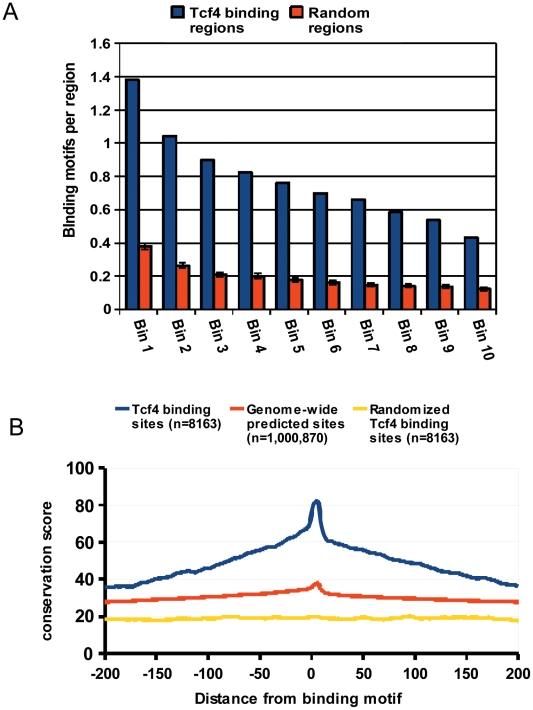
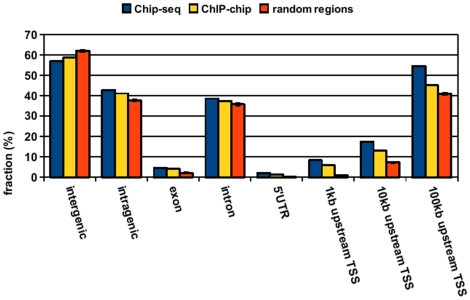

References
-
- Johnson DS, Mortazavi A, Myers RM, Wold B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;316:1497–1502. - PubMed
-
- Robertson G, Hirst M, Bainbridge M, Bilenky M, Zhao Y, et al. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat Methods. 2007;4:651–657. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
