

Neuronal apoptosis induced by endoplasmic reticulum stress is regulated by ATF4-CHOP-mediated induction of the Bcl-2 homology 3-only member PUMA
- PMID: 21159964
- PMCID: PMC6634926
- DOI: 10.1523/JNEUROSCI.1598-10.2010
Neuronal apoptosis induced by endoplasmic reticulum stress is regulated by ATF4-CHOP-mediated induction of the Bcl-2 homology 3-only member PUMA
Abstract
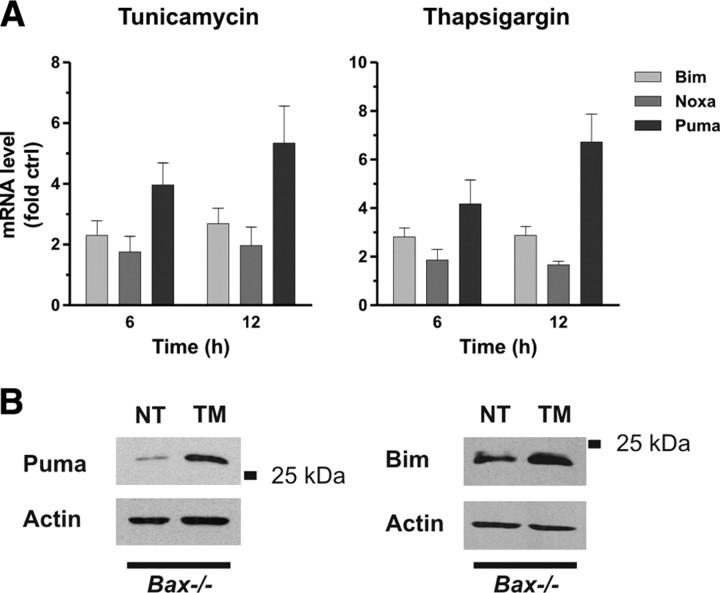
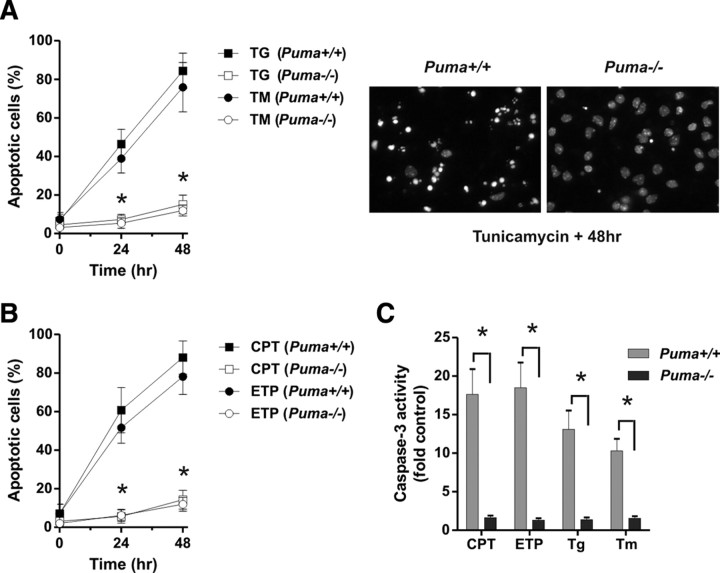
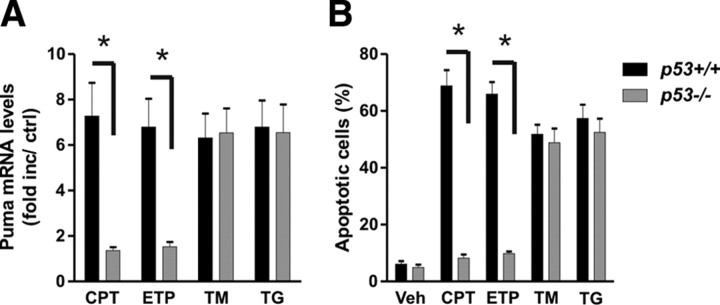
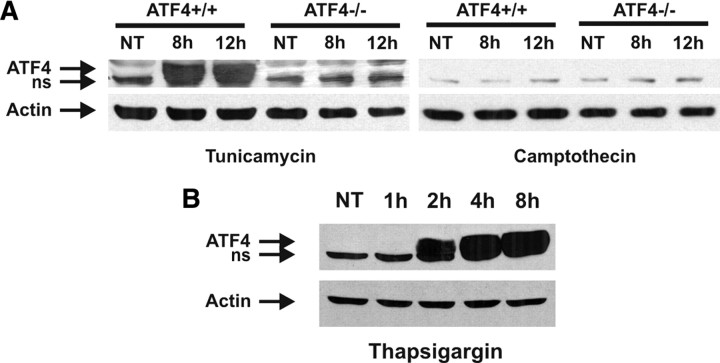
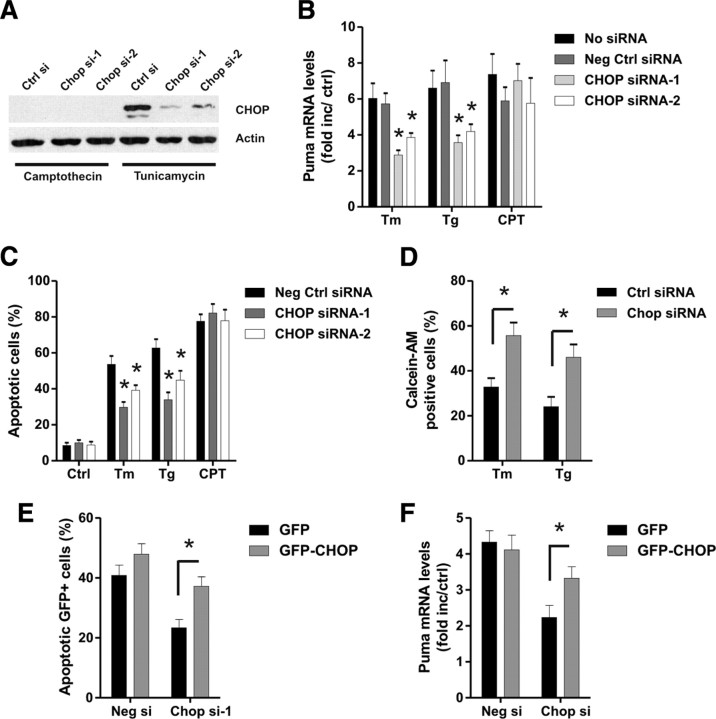
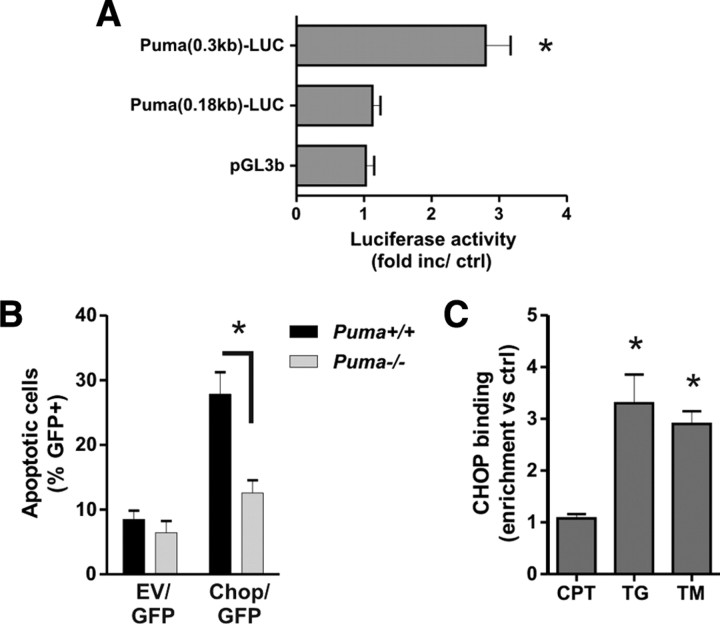
An increasing body of evidence points to a key role of endoplasmic reticulum (ER) stress in acute and chronic neurodegenerative conditions. Extensive ER stress can trigger neuronal apoptosis, but the signaling pathways that regulate this cell death remain unclear. In the present study, we demonstrate that PUMA, a Bcl-2 homology 3 (BH3)-only member of the Bcl-2 family, is transcriptionally activated in cortical neurons by ER stress and is essential for ER-stress-induced cell death. PUMA is known to be a key transcriptional target of p53, but we have found that ER stress triggers PUMA induction and cell death through a p53-independent mechanism mediated by the ER-stress-inducible transcription factor ATF4 (activating transcription factor 4). Specifically, we demonstrate that ectopic expression of ATF4 sensitizes mouse cortical neurons to ER-stress-induced apoptosis and that ATF4-deficient neurons exhibit markedly reduced levels of PUMA expression and cell death. However, chromatin immunoprecipitation experiments suggest that ATF4 does not directly regulate the PUMA promoter. Rather, we found that ATF4 induces expression of the transcription factor CHOP (C/EBP homologous protein) and that CHOP in turn activates PUMA induction. Specifically, we demonstrate that CHOP binds to the PUMA promoter during ER stress and that CHOP knockdown attenuates PUMA induction and neuronal apoptosis. In summary, we have identified a key signaling pathway in ER-stress-induced neuronal death involving ATF4-CHOP-mediated transactivation of the proapoptotic Bcl-2 family member PUMA. We propose that this pathway may be an important therapeutic target relevant to a number of neurodegenerative conditions.
Figures

References
-
- Averous J, Bruhat A, Jousse C, Carraro V, Thiel G, Fafournoux P. Induction of CHOP expression by amino acid limitation requires both ATF4 expression and ATF2 phosphorylation. J Biol Chem. 2004;279:5288–5297. - PubMed
-
- Bouillet P, Strasser A. BH3-only proteins: evolutionarily conserved proapoptotic Bcl-2 family members essential for initiating programmed cell death. J Cell Sci. 2002;115:1567–1574. - PubMed
-
- Bouillet P, Metcalf D, Huang DC, Tarlinton DM, Kay TW, Köntgen F, Adams JM, Strasser A. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science. 1999;286:1735–1738. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous