

CX3CR1 in microglia regulates brain amyloid deposition through selective protofibrillar amyloid-β phagocytosis
- PMID: 21159979
- PMCID: PMC3077120
- DOI: 10.1523/JNEUROSCI.4403-10.2010
CX3CR1 in microglia regulates brain amyloid deposition through selective protofibrillar amyloid-β phagocytosis
Abstract
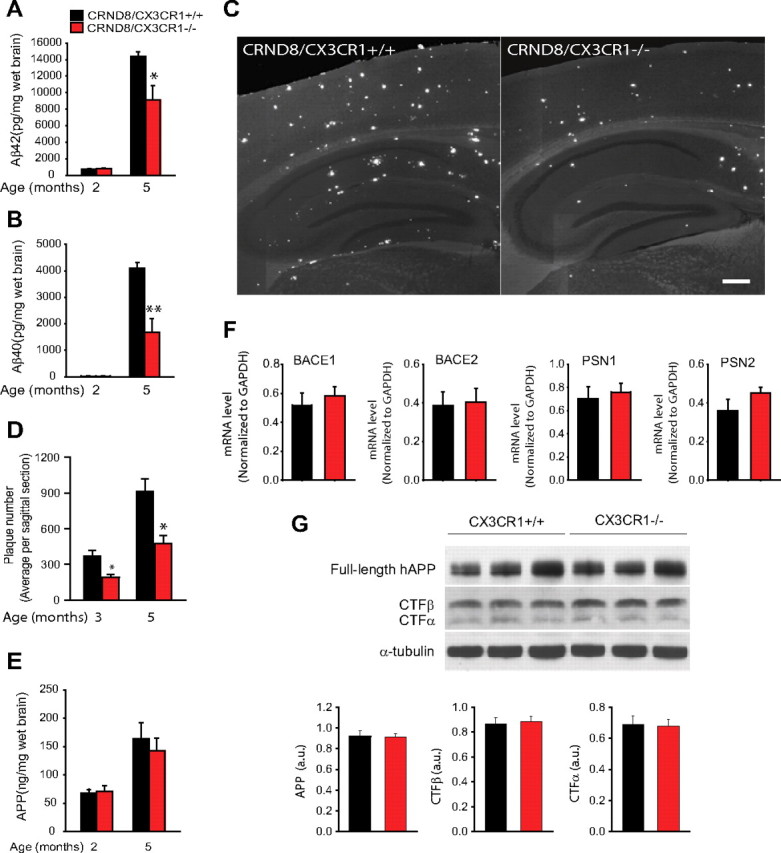
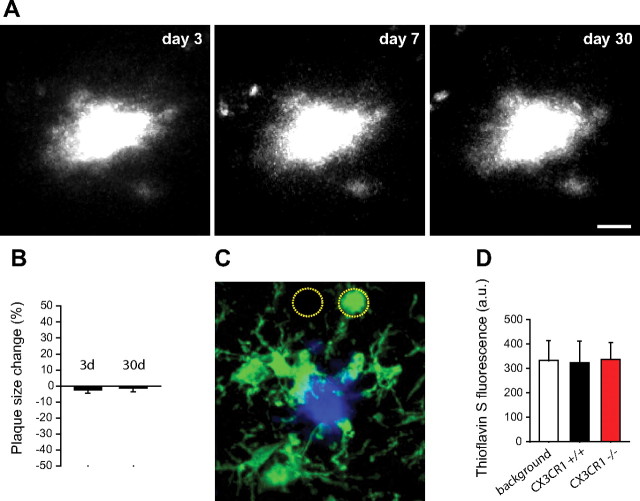
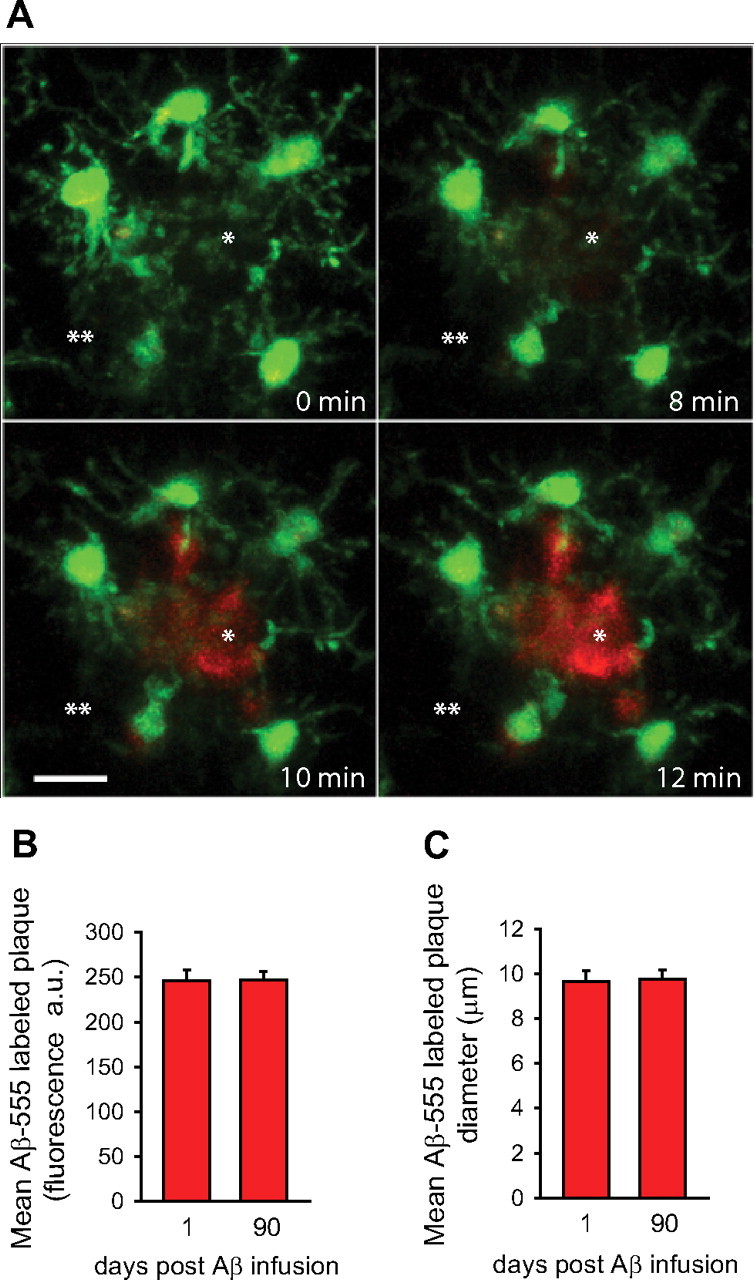
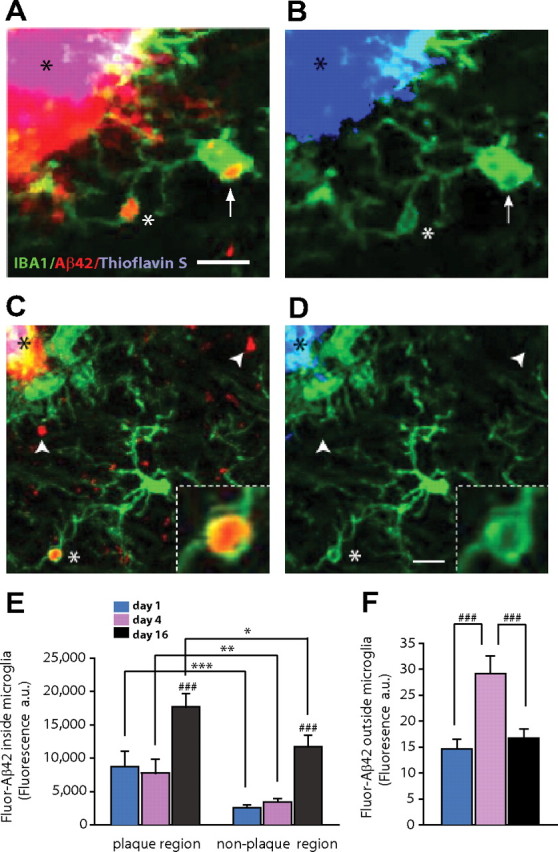
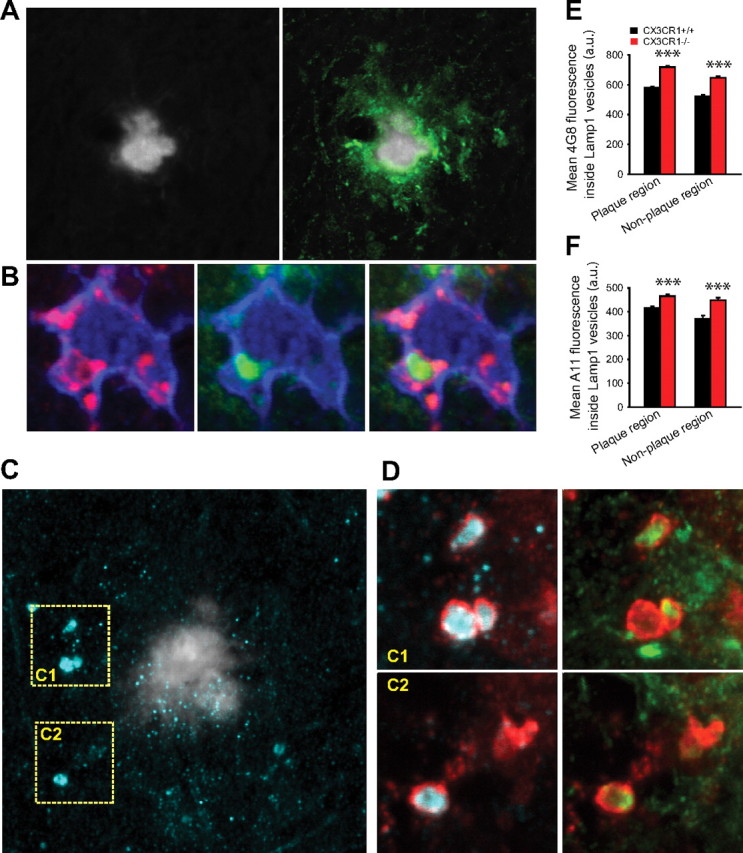
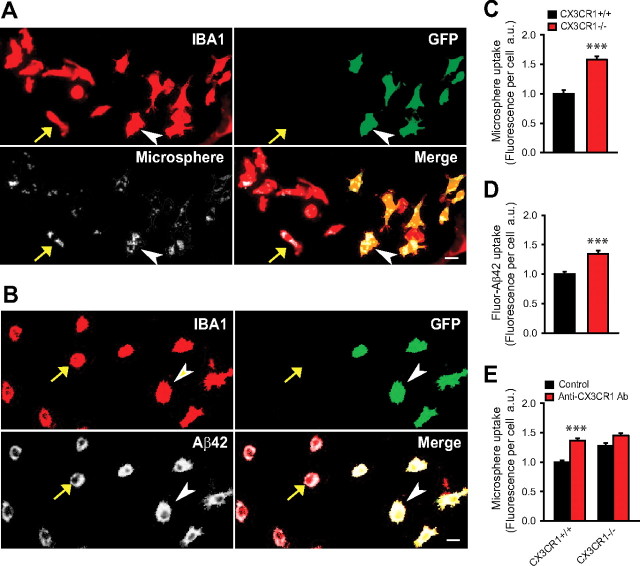
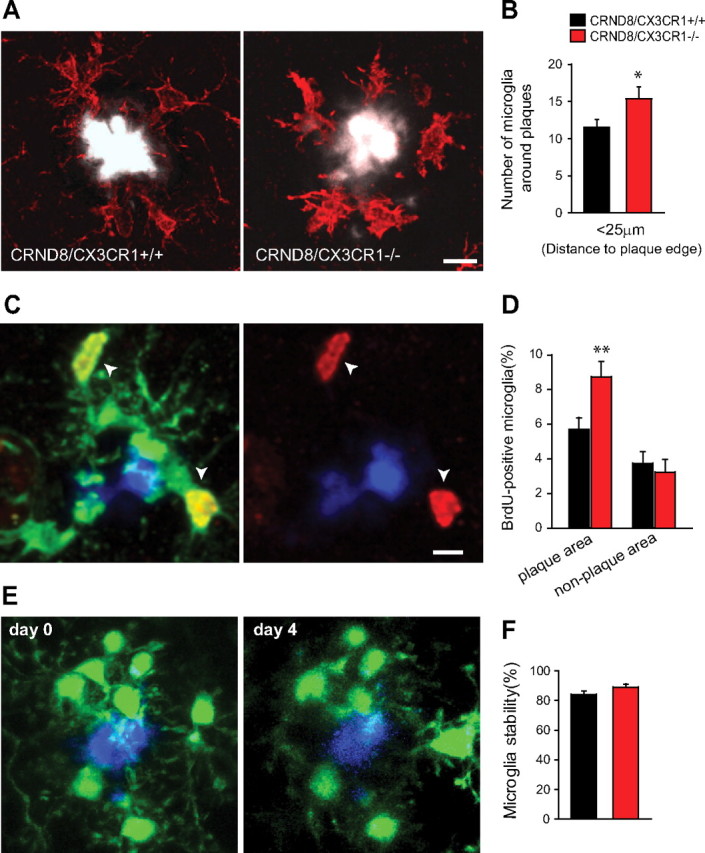
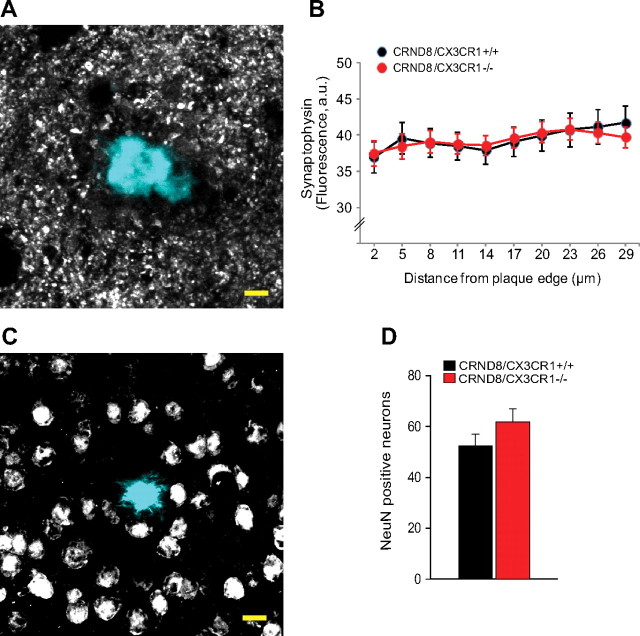
In Alzheimer's disease (AD), amyloid-β (Aβ) deposits are frequently surrounded by activated microglia but the precise role of these cells in disease progression remains unclear. The chemokine receptor CX3CR1 is selectively expressed in microglia and is thought to modulate their activity. To study the specific effects of microglia activation on amyloid pathology in vivo, we crossbred mice lacking CX3CR1 with the Alzheimer's mouse model CRND8. Surprisingly, we found that CX3CR1-deficient mice had lower brain levels of Aβ40 and Aβ42 and reduced amyloid deposits. Quantification of Aβ within microglia and time-lapse two-photon microscopy in live mice revealed that these cells were highly effective at the uptake of protofibrillar amyloid but were incapable of phagocytosis of fibrillar congophilic Aβ. CX3CR1 deletion was associated with increased phagocytic ability, which led to greater amyloid content within microglial phagolysosomes. Furthermore, CX3CR1-deficient mice had an increased number of microglia around individual plaques because of higher proliferative rates, which likely contributed to an overall greater phagocytic capacity. CX3CR1 deletion did not affect the degree of neuronal or synaptic damage around plaques despite increased microglia density. Our results demonstrate that microglia can regulate brain Aβ levels and plaque deposition via selective protofibrillar Aβ phagocytosis. Modulation of microglia activity and proliferation by CX3CR1 signaling may represent a therapeutic strategy for AD.
Figures
References
-
- Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci. 2007;10:1538–1543. - PubMed
-
- Alves-Filho JC, Freitas A, Souto FO, Spiller F, Paula-Neto H, Silva JS, Gazzinelli RT, Teixeira MM, Ferreira SH, Cunha FQ. Regulation of chemokine receptor by Toll-like receptor 2 is critical to neutrophil migration and resistance to polymicrobial sepsis. Proc Natl Acad Sci U S A. 2009;106:4018–4023. - PMC - PubMed
-
- Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases