

Fragmented mitochondria are sensitized to Bax insertion and activation during apoptosis
- PMID: 21160028
- PMCID: PMC3063970
- DOI: 10.1152/ajpcell.00402.2010
Fragmented mitochondria are sensitized to Bax insertion and activation during apoptosis
Abstract
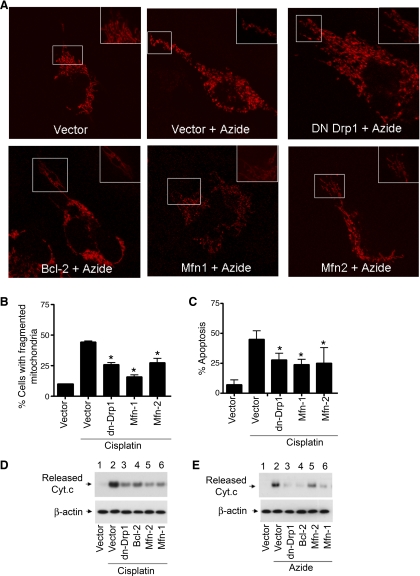
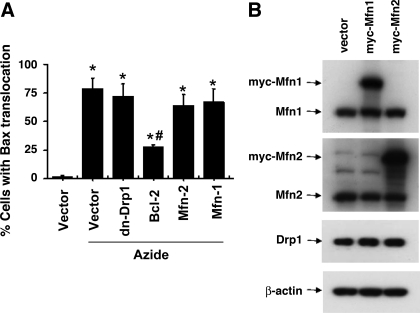
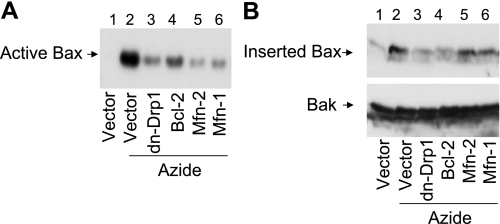
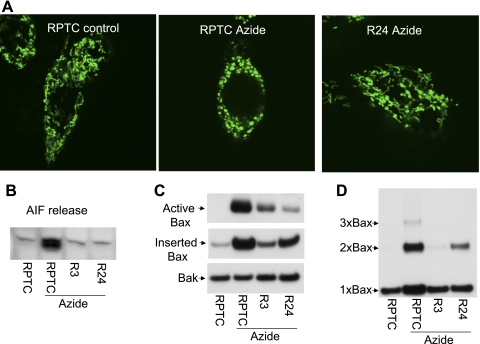
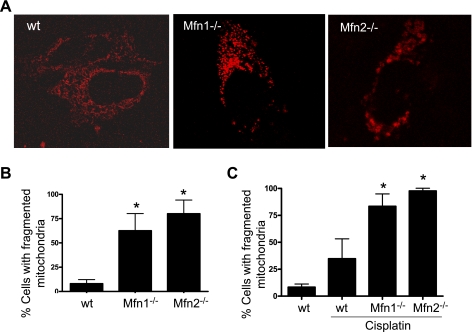
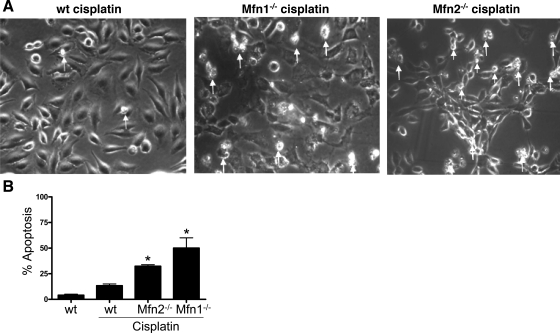
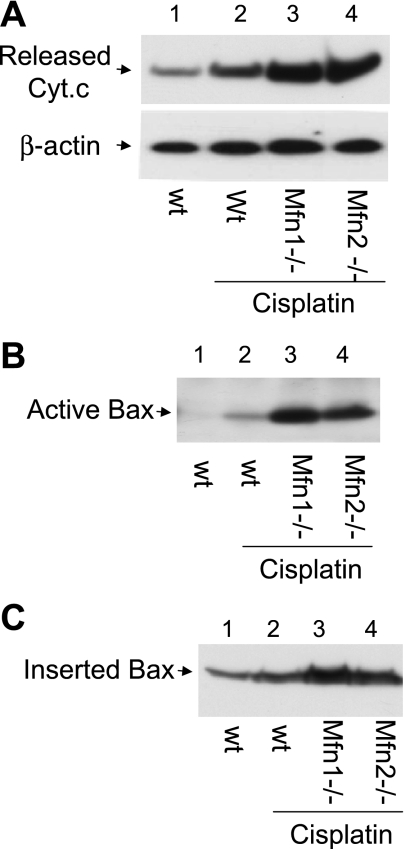
Recent studies have shown mitochondrial fragmentation during cell stress and have suggested a role for the morphological change in mitochondrial injury and ensuing apoptosis. However, the underlying mechanism remains elusive. Here we demonstrate that mitochondrial fragmentation facilitates Bax insertion and activation in mitochondria, resulting in the release of apoptogenic factors. In HeLa cells, overexpression of mitofusins attenuated mitochondrial fragmentation during cisplatin- and azide-induced cell injury, which was accompanied by less apoptosis and less cytochrome c release from mitochondria. Similar effects were shown by inhibiting the mitochondrial fission protein Drp1 with a dominant negative mutant (dn-Drp1). Mitofusins and dn-Drp1 did not seem to significantly affect Bax translocation/accumulation to mitochondria; however, they blocked Bax insertion and activation in mitochondrial membrane. Consistently, in rat kidney proximal tubular cells, small interfering RNA knockdown of Drp1 prevented mitochondrial fragmentation during azide-induced ATP depletion, which was accompanied by less Bax activation, insertion, and oligomerization in mitochondria. These cells released less cytochrome c and AIF from mitochondria and showed significantly lower apoptosis. Finally, mitofusin-null mouse embryonic fibroblasts (MEF) had fragmented mitochondria. These MEFs were more sensitive to cisplatin-induced Bax activation, release of cytochrome c, and apoptosis. Together, this study provides further support for a role of mitochondrial fragmentation in mitochondrial injury and apoptosis. Mechanistically, mitochondrial fragmentation may sensitize the cells to Bax insertion and activation in mitochondria, facilitating the release of apoptogenic factors and consequent apoptosis.
Figures
References
-
- Arnoult D, Rismanchi N, Grodet A, Roberts RG, Seeburg DP, Estaquier J, Sheng M, Blackstone C. Bax/Bak-dependent release of DDP/TIMM8a promotes Drp1-mediated mitochondrial fission and mitoptosis during programmed cell death. Curr Biol 15: 2112–2118, 2005 - PubMed
-
- Brooks C, Dong Z. Regulation of mitochondrial morphological dynamics during apoptosis by Bcl-2 family proteins: a key in Bak? Cell Cycle 6: 3043–3047, 2007 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous