

A novel strategy for NMR resonance assignment and protein structure determination
- PMID: 21161328
- PMCID: PMC3715383
- DOI: 10.1007/s10858-010-9458-0
A novel strategy for NMR resonance assignment and protein structure determination
Abstract
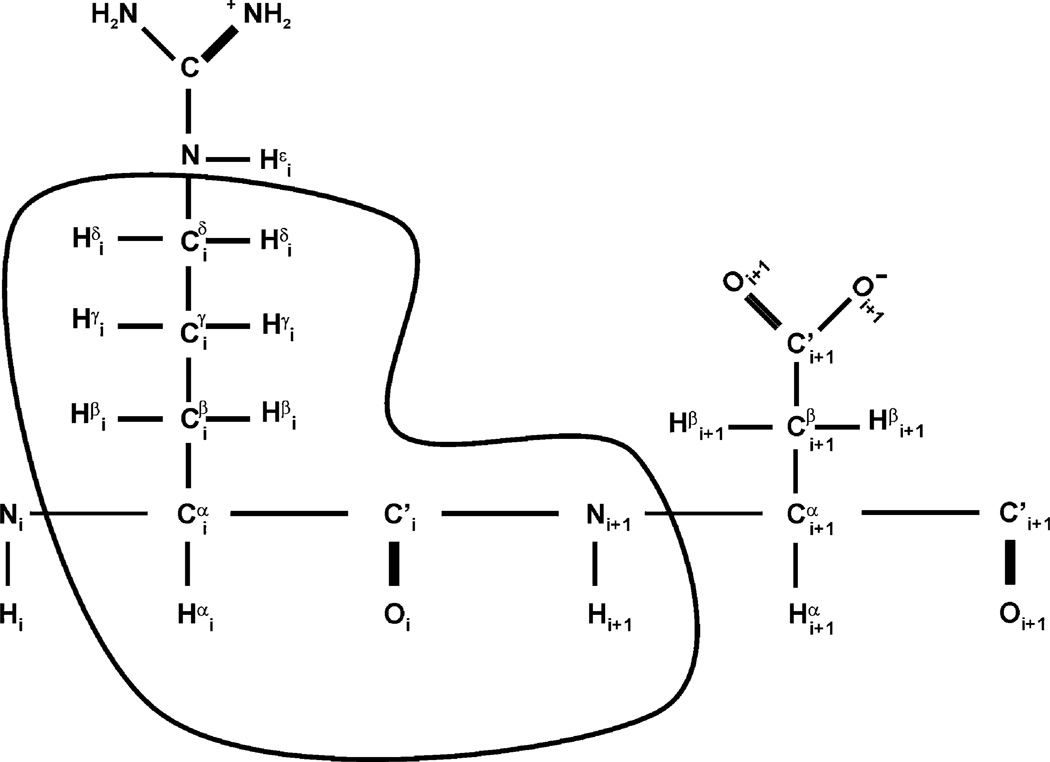
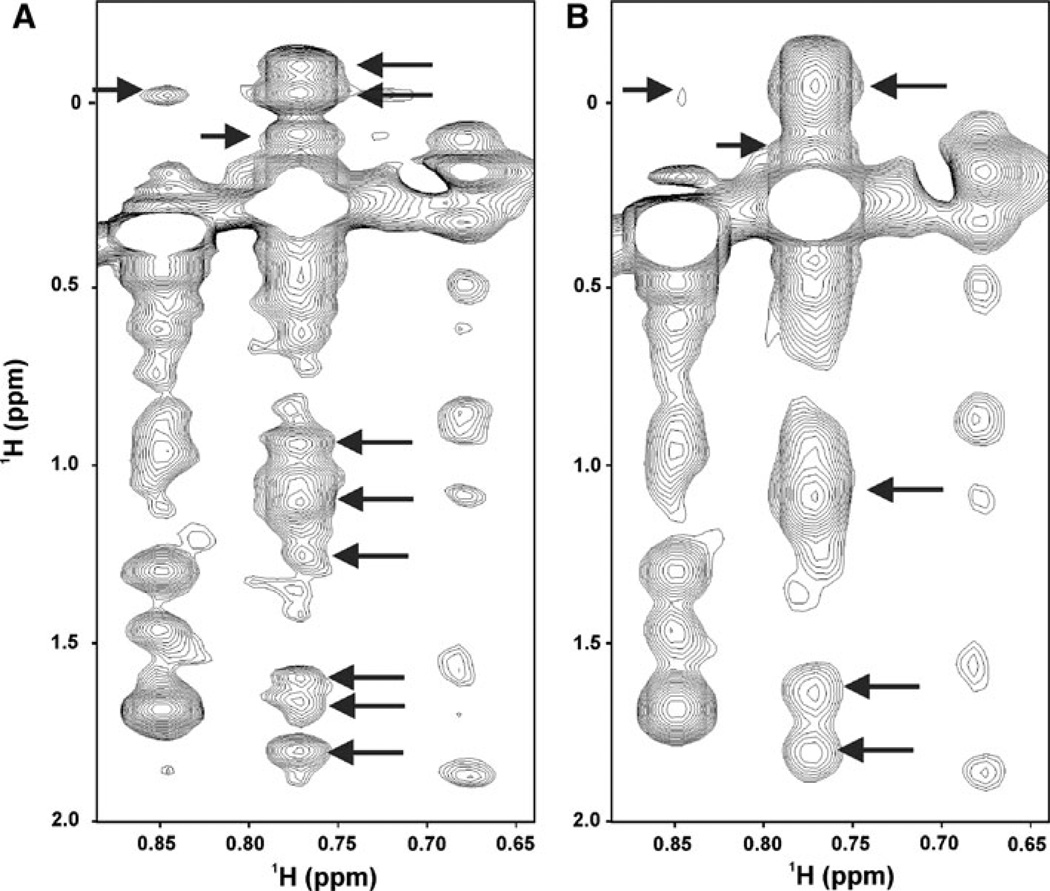
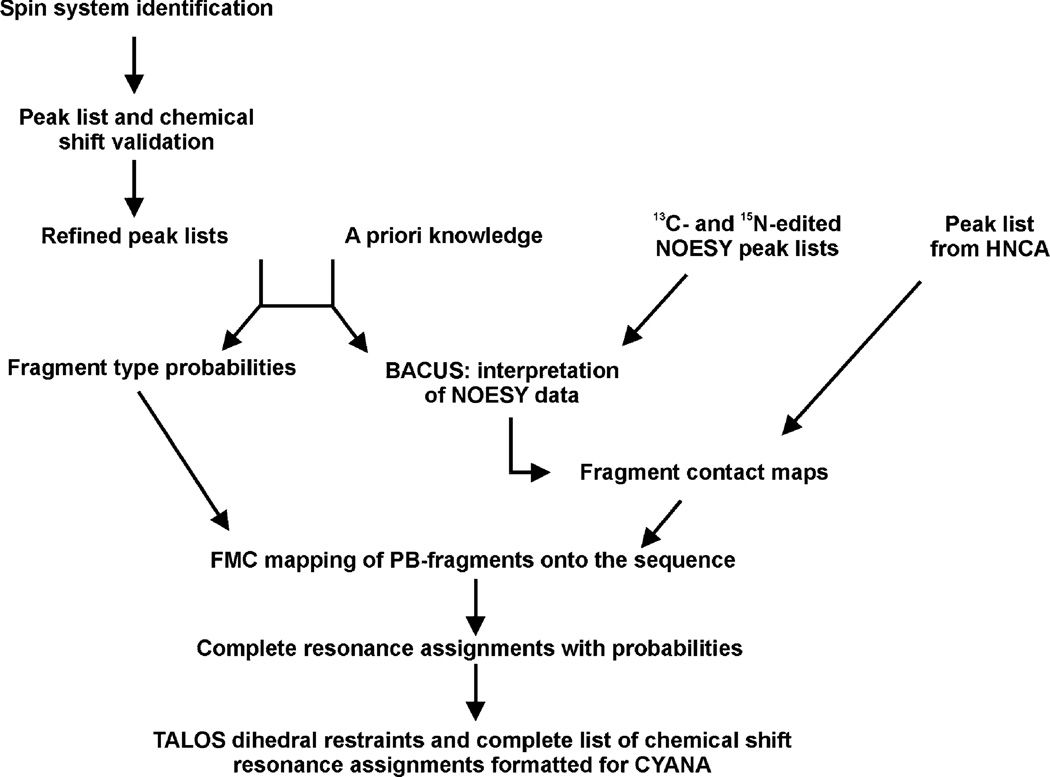
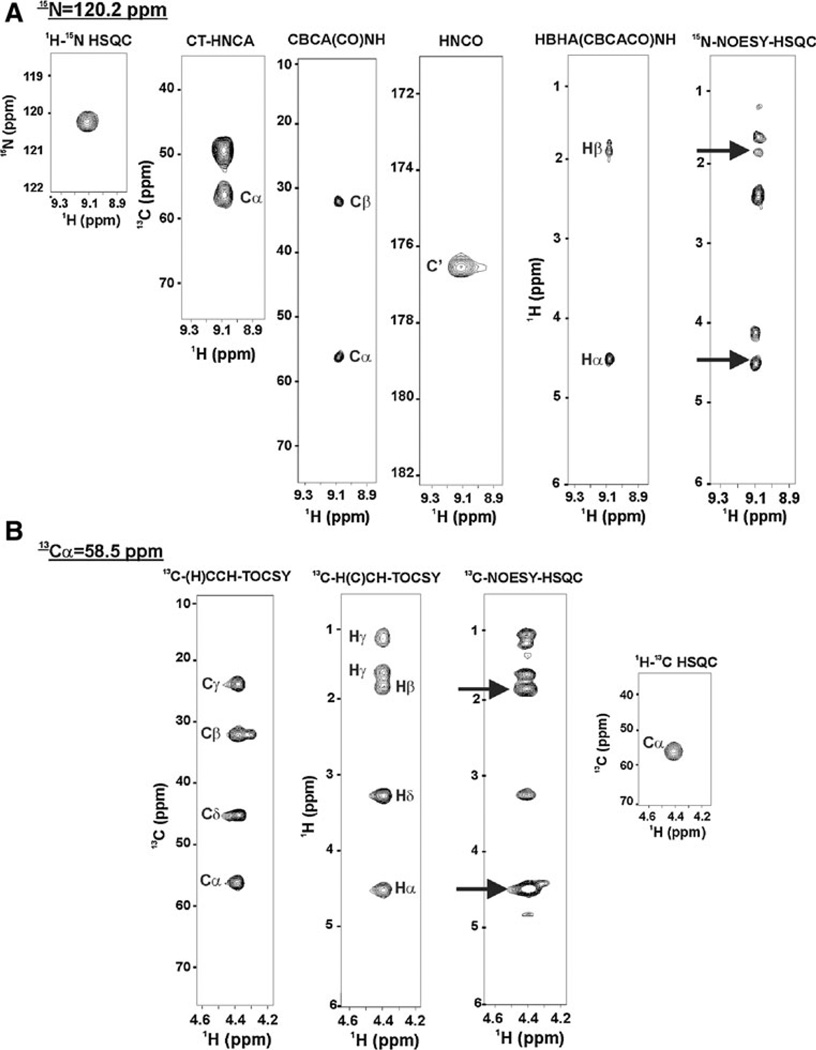
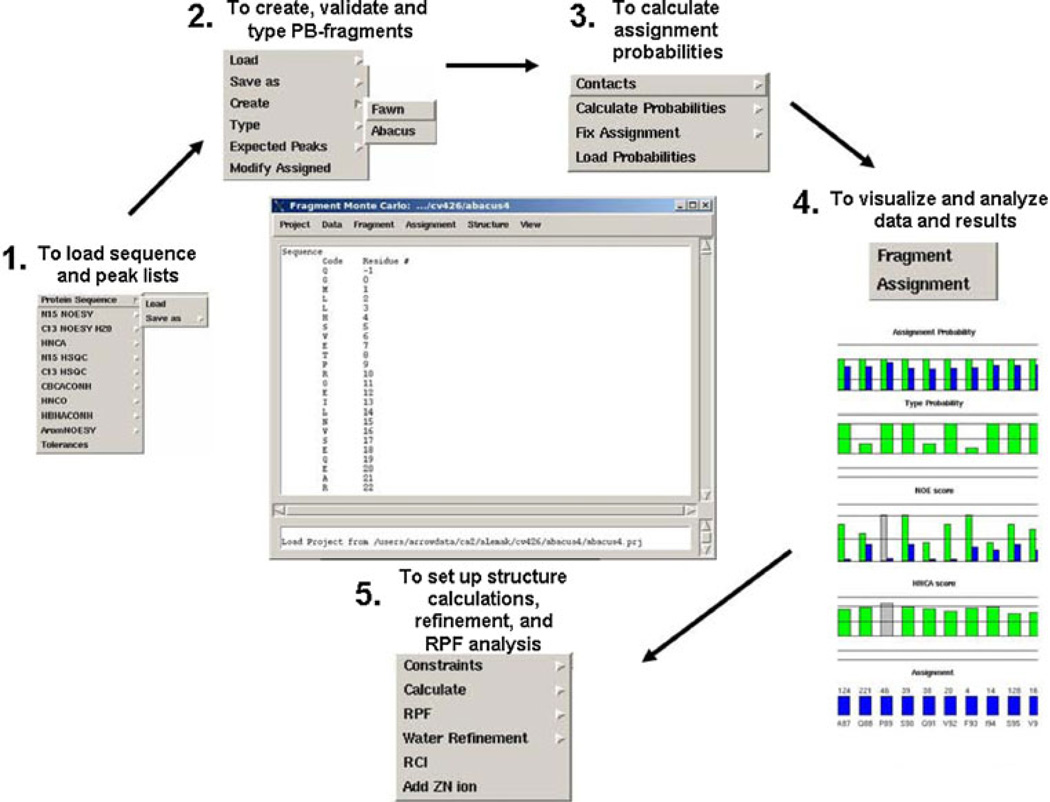
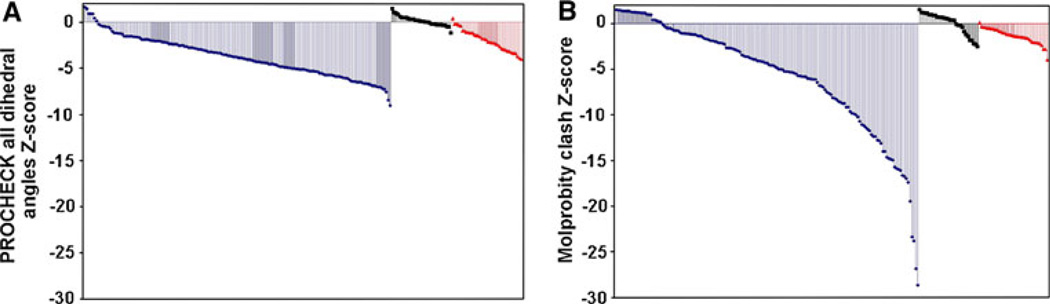
The quality of protein structures determined by nuclear magnetic resonance (NMR) spectroscopy is contingent on the number and quality of experimentally-derived resonance assignments, distance and angular restraints. Two key features of protein NMR data have posed challenges for the routine and automated structure determination of small to medium sized proteins; (1) spectral resolution - especially of crowded nuclear Overhauser effect spectroscopy (NOESY) spectra, and (2) the reliance on a continuous network of weak scalar couplings as part of most common assignment protocols. In order to facilitate NMR structure determination, we developed a semi-automated strategy that utilizes non-uniform sampling (NUS) and multidimensional decomposition (MDD) for optimal data collection and processing of selected, high resolution multidimensional NMR experiments, combined it with an ABACUS protocol for sequential and side chain resonance assignments, and streamlined this procedure to execute structure and refinement calculations in CYANA and CNS, respectively. Two graphical user interfaces (GUIs) were developed to facilitate efficient analysis and compilation of the data and to guide automated structure determination. This integrated method was implemented and refined on over 30 high quality structures of proteins ranging from 5.5 to 16.5 kDa in size.
Figures
References
-
- Atreya H, Sahu SC, Chary KV, Govil G. A tracked approach for automated NMR assignments in proteins (TATAPRO) J Biomol NMR. 2000;17:125–136. - PubMed
-
- Barna J, Laue ED. Conventional and exponential sampling for 2D NMR experiments with application to a 2D NMR spectrum of a protein. J Magn Reson. 1987;75:387–389.
-
- Bax A, Clore GM, Gronenborn AM. 1H-1H correlation via isotropic mixing of 13C magnetization: a new three-dimensional approach for assigning 1H and 13C spectra of 13C-enriched proteins. J Magn Reson B. 1990;88:425–431.
-
- Bhattacharya A, Tejero R, Montelione GT. Evaluating protein structures deterined by structural genomics consortia. Proteins. 2007;66:778–795. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
