

Inference of hierarchical regulatory network of estrogen-dependent breast cancer through ChIP-based data
- PMID: 21167036
- PMCID: PMC3012048
- DOI: 10.1186/1752-0509-4-170
Inference of hierarchical regulatory network of estrogen-dependent breast cancer through ChIP-based data
Abstract
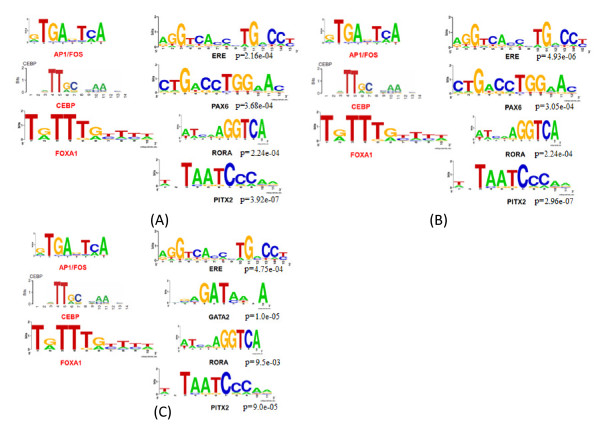
Background: Global profiling of in vivo protein-DNA interactions using ChIP-based technologies has evolved rapidly in recent years. Although many genome-wide studies have identified thousands of ERα binding sites and have revealed the associated transcription factor (TF) partners, such as AP1, FOXA1 and CEBP, little is known about ERα associated hierarchical transcriptional regulatory networks.
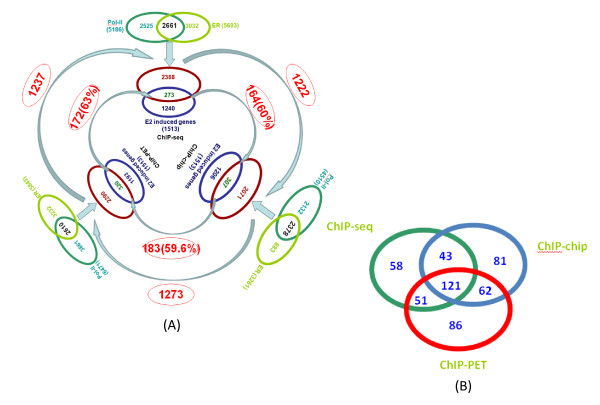
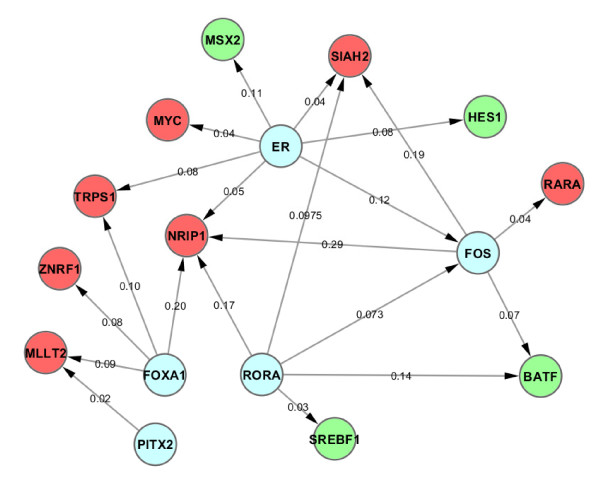
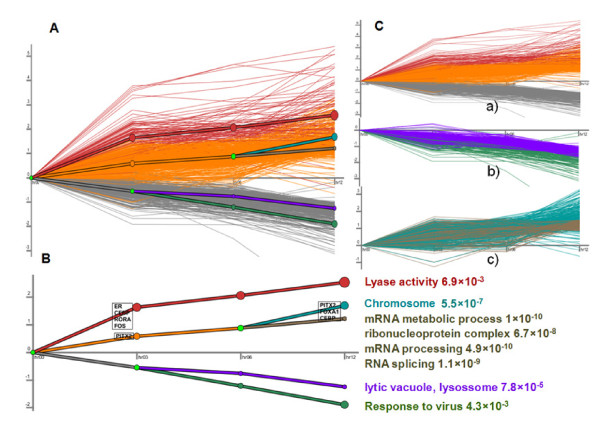
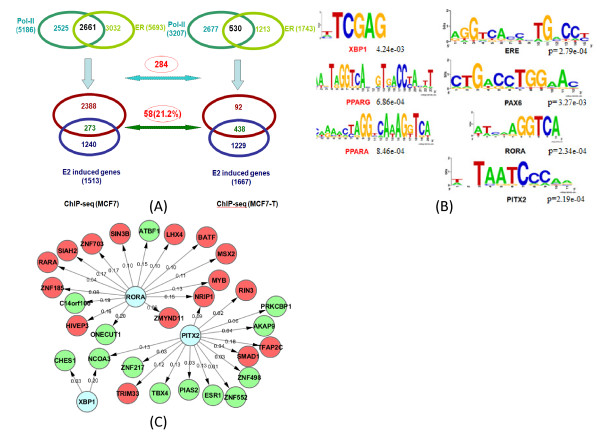
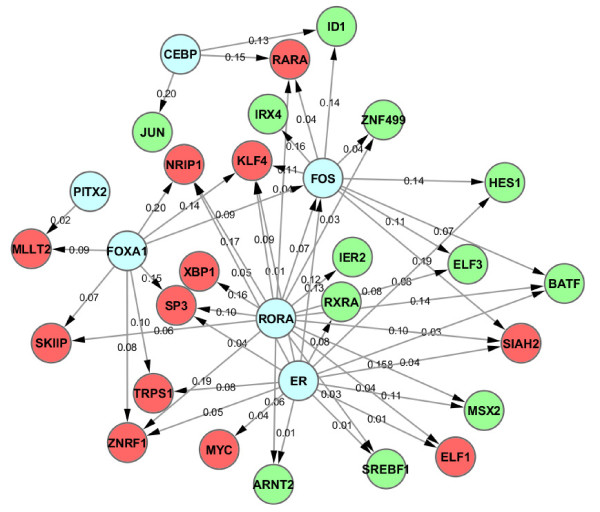
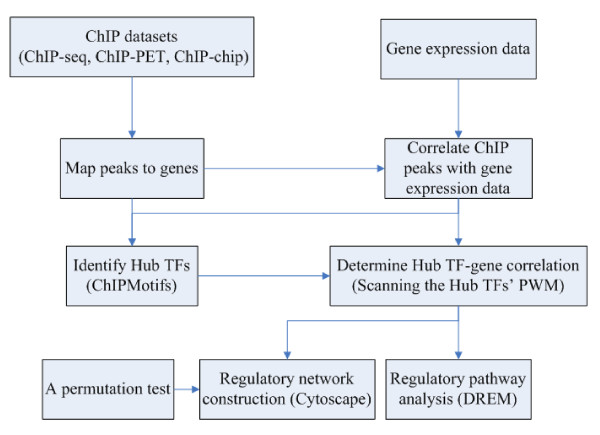
Results: In this study, we applied computational approaches to analyze three public available ChIP-based datasets: ChIP-seq, ChIP-PET and ChIP-chip, and to investigate the hierarchical regulatory network for ERα and ERα partner TFs regulation in estrogen-dependent breast cancer MCF7 cells. 16 common TFs and two common new TF partners (RORA and PITX2) were found among ChIP-seq, ChIP-chip and ChIP-PET datasets. The regulatory networks were constructed by scanning the ChIP-peak region with TF specific position weight matrix (PWM). A permutation test was performed to test the reliability of each connection of the network. We then used DREM software to perform gene ontology function analysis on the common genes. We found that FOS, PITX2, RORA and FOXA1 were involved in the up-regulated genes.We also conducted the ERα and Pol-II ChIP-seq experiments in tamoxifen resistance MCF7 cells (denoted as MCF7-T in this study) and compared the difference between MCF7 and MCF7-T cells. The result showed very little overlap between these two cells in terms of targeted genes (21.2% of common genes) and targeted TFs (25% of common TFs). The significant dissimilarity may indicate totally different transcriptional regulatory mechanisms between these two cancer cells.
Conclusions: Our study uncovers new estrogen-mediated regulatory networks by mining three ChIP-based data in MCF7 cells and ChIP-seq data in MCF7-T cells. We compared the different ChIP-based technologies as well as different breast cancer cells. Our computational analytical approach may guide biologists to further study the underlying mechanisms in breast cancer cells or other human diseases.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
