

Nuclear factors involved in mitochondrial translation cause a subgroup of combined respiratory chain deficiency
- PMID: 21169334
- PMCID: PMC3707321
- DOI: 10.1093/brain/awq320
Nuclear factors involved in mitochondrial translation cause a subgroup of combined respiratory chain deficiency
Abstract
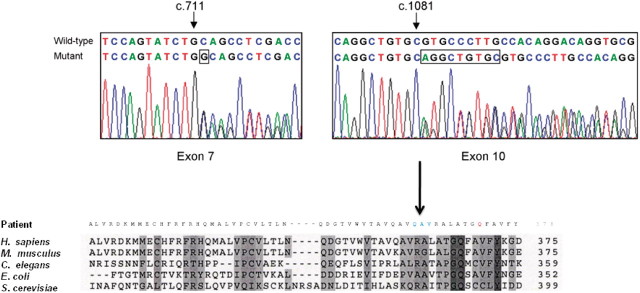
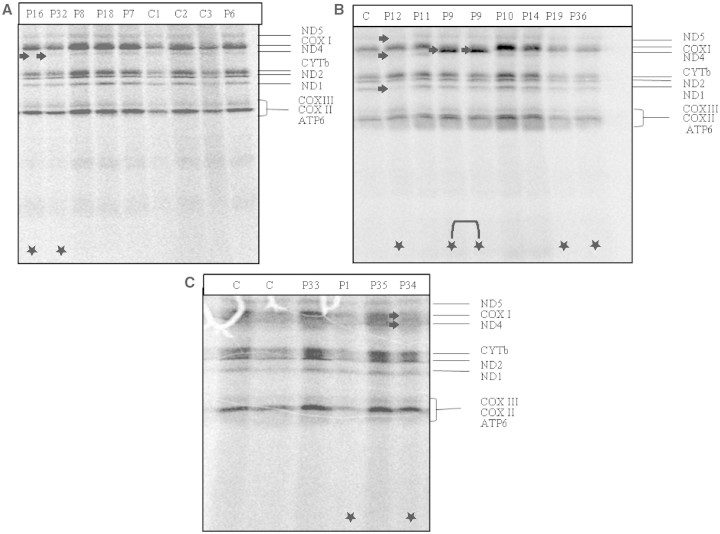
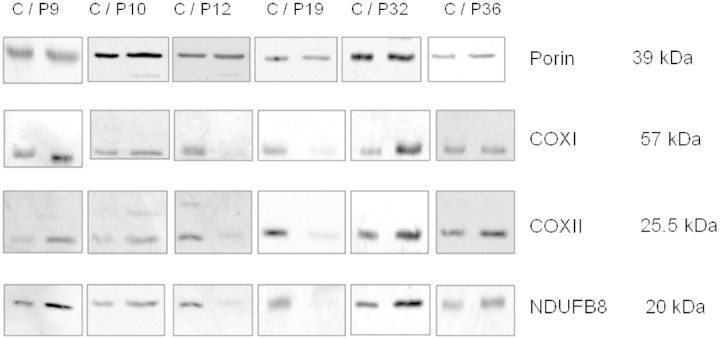
Mutations in several mitochondrial DNA and nuclear genes involved in mitochondrial protein synthesis have recently been reported in combined respiratory chain deficiency, indicating a generalized defect in mitochondrial translation. However, the number of patients with pathogenic mutations is small, implying that nuclear defects of mitochondrial translation are either underdiagnosed or intrauterine lethal. No comprehensive studies have been reported on large cohorts of patients with combined respiratory chain deficiency addressing the role of nuclear genes affecting mitochondrial protein synthesis to date. We investigated a cohort of 52 patients with combined respiratory chain deficiency without causative mitochondrial DNA mutations, rearrangements or depletion, to determine whether a defect in mitochondrial translation defines the pathomechanism of their clinical disease. We followed a combined approach of sequencing known nuclear genes involved in mitochondrial protein synthesis (EFG1, EFTu, EFTs, MRPS16, TRMU), as well as performing in vitro functional studies in 22 patient cell lines. The majority of our patients were children (<15 years), with an early onset of symptoms <1 year of age (65%). The most frequent clinical presentation was mitochondrial encephalomyopathy (63%); however, a number of patients showed cardiomyopathy (33%), isolated myopathy (15%) or hepatopathy (13%). Genomic sequencing revealed compound heterozygous mutations in the mitochondrial transfer ribonucleic acid modifying factor (TRMU) in a single patient only, presenting with early onset, reversible liver disease. No pathogenic mutation was detected in any of the remaining 51 patients in the other genes analysed. In vivo labelling of mitochondrial polypeptides in 22 patient cell lines showed overall (three patients) or selective (four patients) defects of mitochondrial translation. Immunoblotting for mitochondrial proteins revealed decreased steady state levels of proteins in some patients, but normal or increased levels in others, indicating a possible compensatory mechanism. In summary, candidate gene sequencing in this group of patients has a very low detection rate (1/52), although in vivo labelling of mitochondrial translation in 22 patient cell lines indicate that a nuclear defect affecting mitochondrial protein synthesis is responsible for about one-third of combined respiratory chain deficiencies (7/22). In the remaining patients, the impaired respiratory chain activity is most likely the consequence of several different events downstream of mitochondrial translation. Clinical classification of patients with biochemical analysis, genetic testing and, more importantly, in vivo labelling and immunoblotting of mitochondrial proteins show incoherent results, but a systematic review of these data in more patients may reveal underlying mechanisms, and facilitate the identification of novel factors involved in combined respiratory chain deficiency.
Figures
References
-
- Andrews RM, Kubacka I, Chinnery PF, Lightowlers RN, Turnbull DM, Howell N. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat Genet. 1999;23:147. - PubMed
-
- Antonicka H, Sasarman F, Kennaway NG, Shoubridge EA. The molecular basis for tissue specificity of the oxidative phosphorylation deficiencies in patients with mutations in the mitochondrial translation factor EFG1. Hum Mol Genet. 2006;15:1835–46. - PubMed
-
- Barthélémy C, Ogier de Baulny H, Diaz J, Cheval MA, Frachon P, et al. Late-onset mitochondrial DNA depletion: DNA copy number, multiple deletions, and compensation. Ann Neurol. 2001;49:607–17. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
