

Haplotype-resolved genome sequencing of a Gujarati Indian individual
- PMID: 21170042
- PMCID: PMC3116788
- DOI: 10.1038/nbt.1740
Haplotype-resolved genome sequencing of a Gujarati Indian individual
Erratum in
- Nat Biotechnol. 2011 May;29(5):459
Abstract
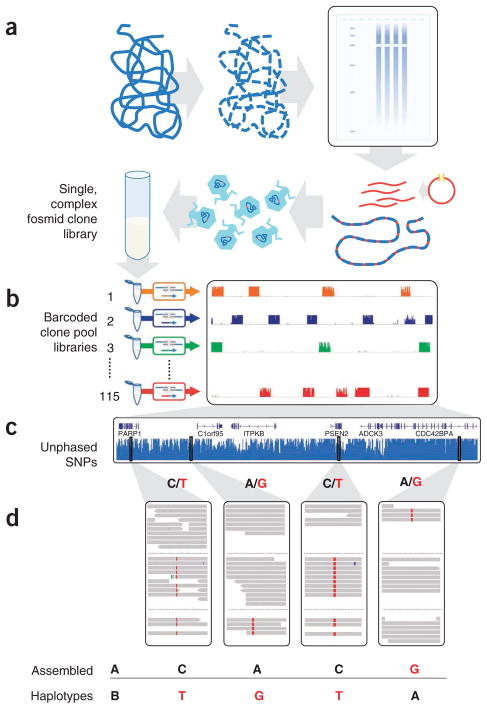
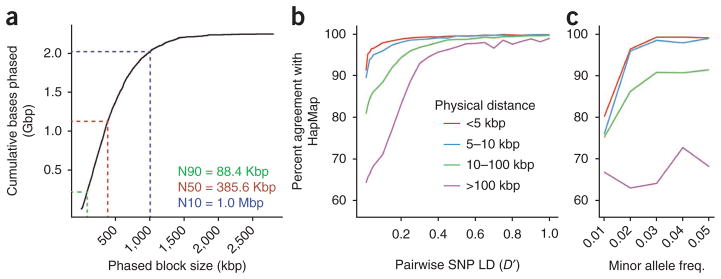
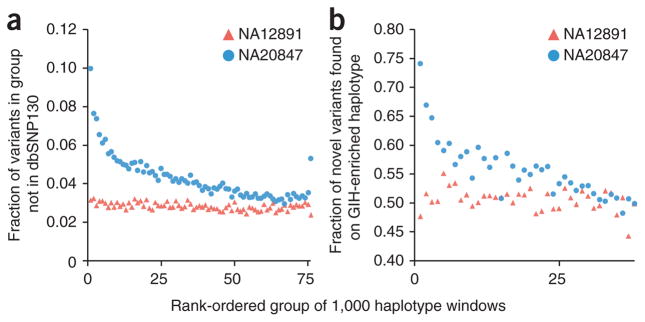
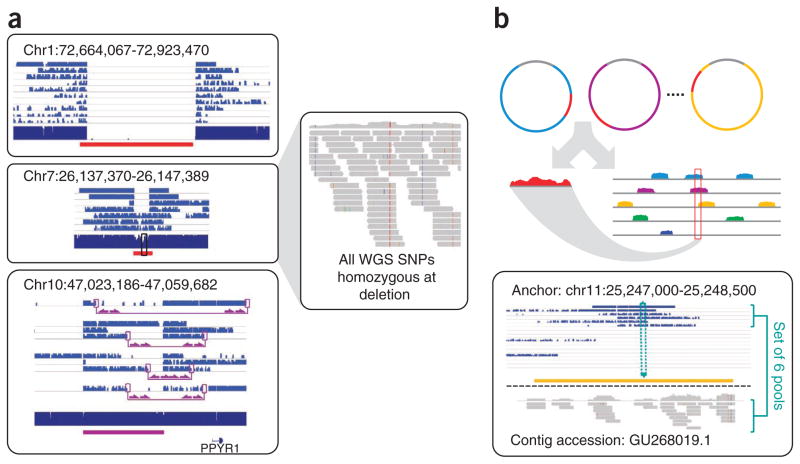
Haplotype information is essential to the complete description and interpretation of genomes, genetic diversity and genetic ancestry. Although individual human genome sequencing is increasingly routine, nearly all such genomes are unresolved with respect to haplotype. Here we combine the throughput of massively parallel sequencing with the contiguity information provided by large-insert cloning to experimentally determine the haplotype-resolved genome of a South Asian individual. A single fosmid library was split into a modest number of pools, each providing ∼3% physical coverage of the diploid genome. Sequencing of each pool yielded reads overwhelmingly derived from only one homologous chromosome at any given location. These data were combined with whole-genome shotgun sequence to directly phase 94% of ascertained heterozygous single nucleotide polymorphisms (SNPs) into long haplotype blocks (N50 of 386 kilobases (kbp)). This method also facilitates the analysis of structural variation, for example, to anchor novel insertions to specific locations and haplotypes.
Conflict of interest statement
The authors declare competing financial interests: details accompany the full-text HTML version of the paper at
Figures
Comment in
-
Genomics: No half measures for haplotypes.Nat Rev Genet. 2011 Feb;12(2):77. doi: 10.1038/nrg2939. Epub 2010 Dec 30. Nat Rev Genet. 2011. PMID: 21191422 No abstract available.
-
The next phase in human genetics.Nat Biotechnol. 2011 Jan;29(1):38-9. doi: 10.1038/nbt.1757. Nat Biotechnol. 2011. PMID: 21221098 No abstract available.
-
One genome, two haplotypes.Nat Methods. 2011 Feb;8(2):107. doi: 10.1038/nmeth0211-107. Nat Methods. 2011. PMID: 21355116 No abstract available.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
