

Genome-wide identification of polycomb-associated RNAs by RIP-seq
- PMID: 21172659
- PMCID: PMC3021903
- DOI: 10.1016/j.molcel.2010.12.011
Genome-wide identification of polycomb-associated RNAs by RIP-seq
Abstract
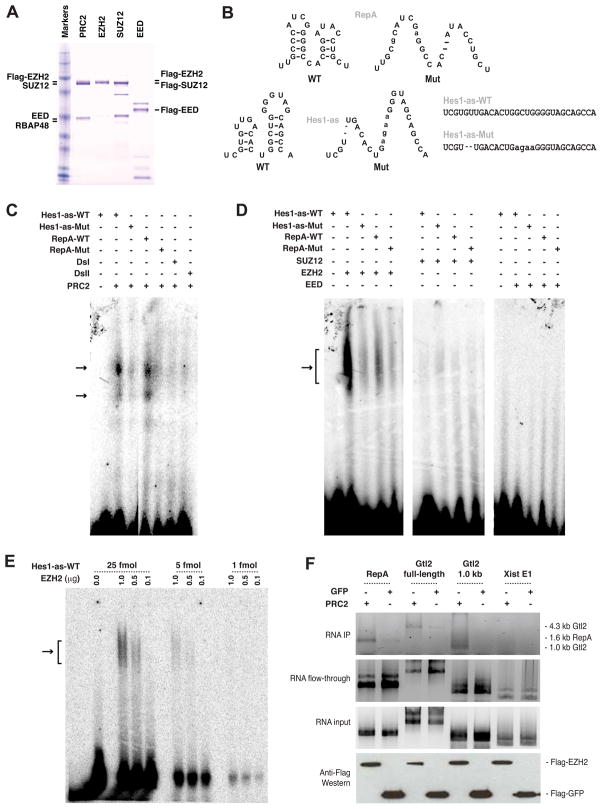
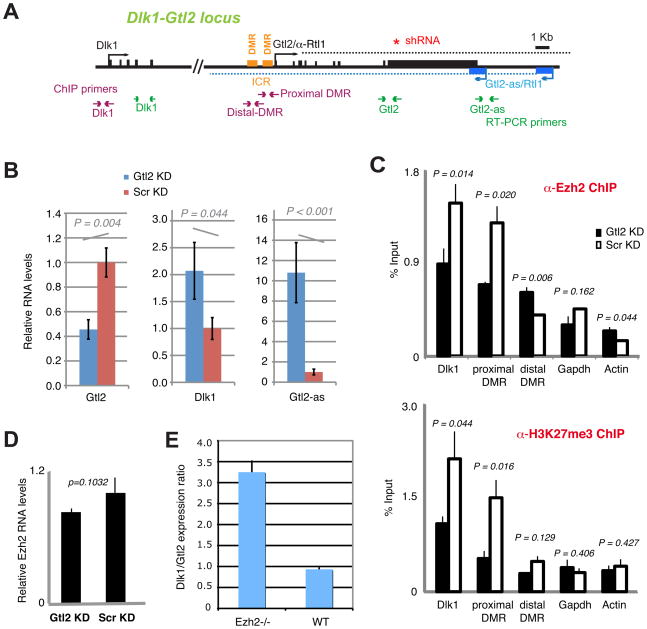
Polycomb proteins play essential roles in stem cell renewal and human disease. Recent studies of HOX genes and X inactivation have provided evidence for RNA cofactors in Polycomb repressive complex 2 (PRC2). Here we develop a RIP-seq method to capture the PRC2 transcriptome and identify a genome-wide pool of >9000 PRC2-interacting RNAs in embryonic stem cells. The transcriptome includes antisense, intergenic, and promoter-associated transcripts, as well as many unannotated RNAs. A large number of transcripts occur within imprinted regions, oncogene and tumor suppressor loci, and stem cell-related bivalent domains. We provide evidence for direct RNA-protein interactions, most likely via the Ezh2 subunit. We also identify Gtl2 RNA as a PRC2 cofactor that directs PRC2 to the reciprocally imprinted Dlk1 coding gene. Thus, Polycomb proteins interact with a genome-wide family of RNAs, some of which may be used as biomarkers and therapeutic targets for human disease.
Copyright © 2010 Elsevier Inc. All rights reserved.
Figures
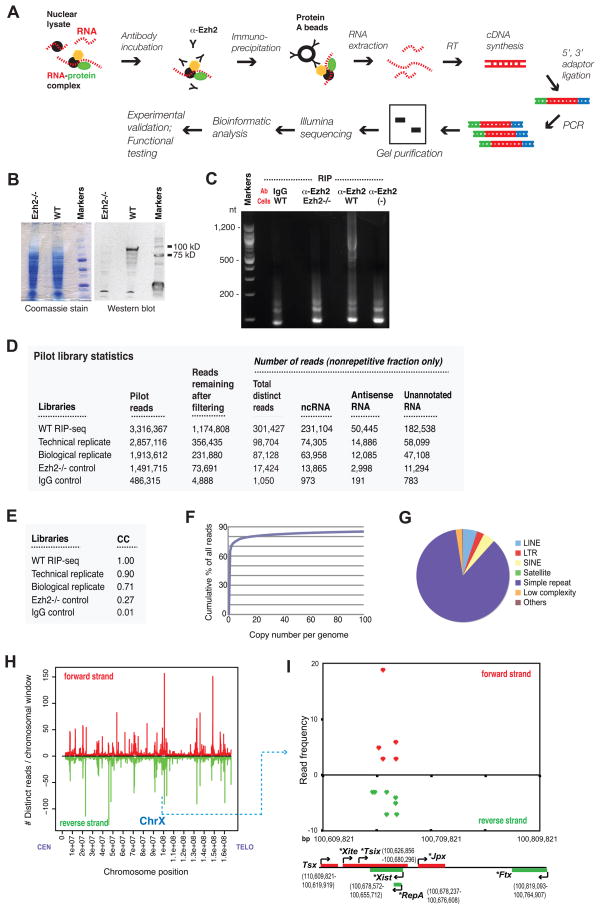
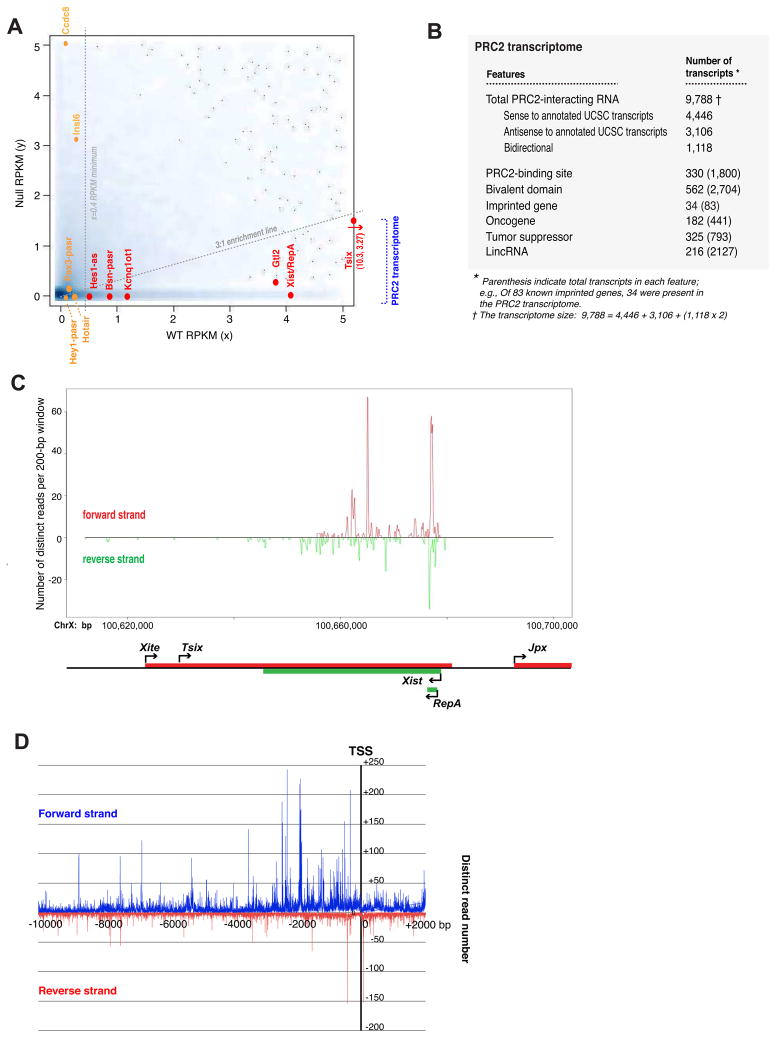
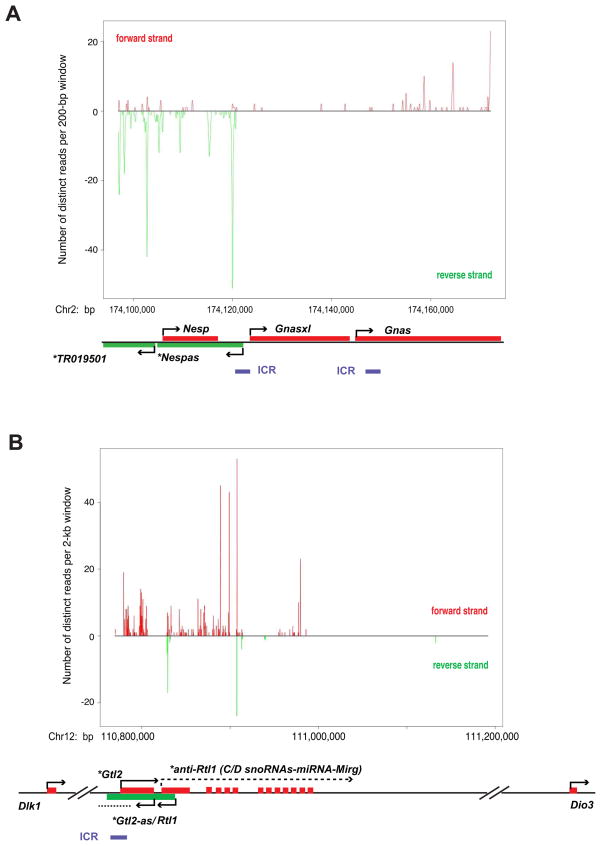
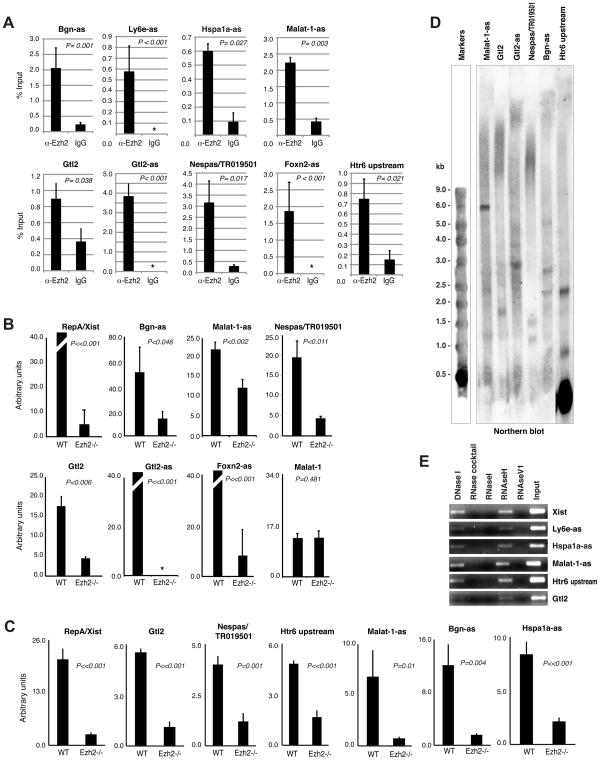
References
-
- Axelson H. The Notch signaling cascade in neuroblastoma: role of the basic helix-loop-helix proteins HASH-1 and HES-1. Cancer Lett. 2004;204:171–178. - PubMed
-
- Bernardi R, Pandolfi PP. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat Rev Mol Cell Biol. 2007;8:1006–1016. - PubMed
-
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006a;125:315–326. - PubMed
-
- Bernstein E, Allis CD. RNA meets chromatin. Genes Dev. 2005;19:1635–1655. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
