

RNA sequencing reveals the role of splicing polymorphisms in regulating human gene expression
- PMID: 21173033
- PMCID: PMC3065702
- DOI: 10.1101/gr.111211.110
RNA sequencing reveals the role of splicing polymorphisms in regulating human gene expression
Abstract
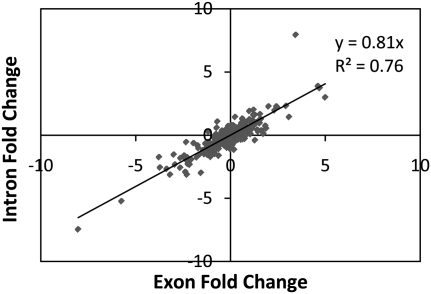
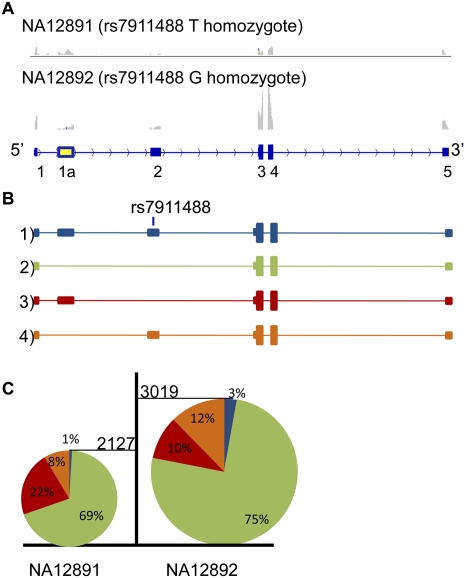
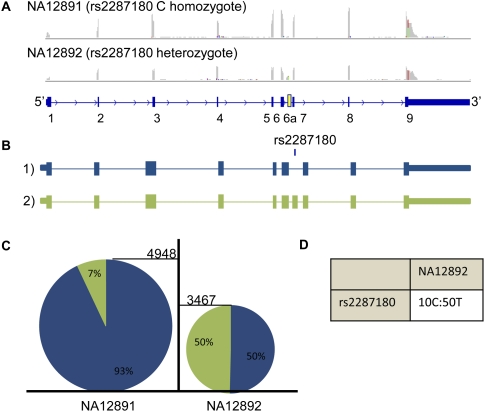
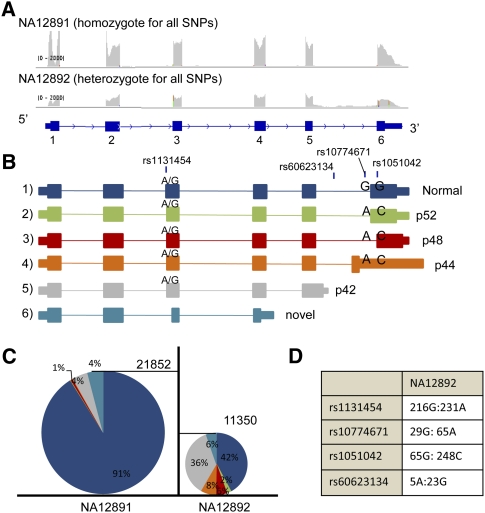
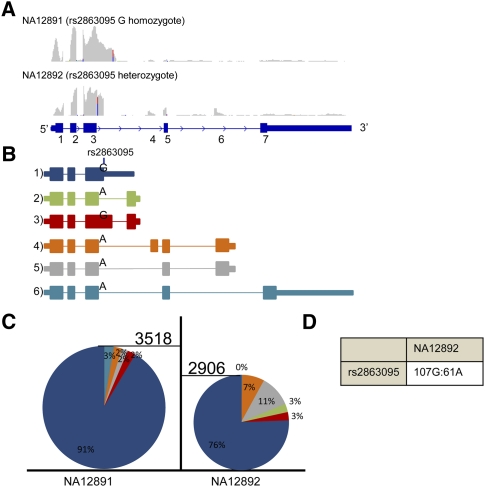
Expression levels of many human genes are under the genetic control of expression quantitative trait loci (eQTLs). Despite technological advances, the precise molecular mechanisms underlying most eQTLs remain elusive. Here, we use deep mRNA sequencing of two CEU individuals to investigate those mechanisms, with particular focus on the role of splicing control loci (sQTLs). We identify a large number of genes that are differentially spliced between the two samples and associate many of those differences with nearby single nucleotide polymorphisms (SNPs). Subsequently, we investigate the potential effect of splicing SNPs on eQTL control in general. We find a significant enrichment of alternative splicing (AS) events within a set of highly confident eQTL targets discovered in previous studies, suggesting a role of AS in regulating overall gene expression levels. Next, we demonstrate high correlation between the levels of mature (exonic) and unprocessed (intronic) RNA, implying that ∼75% of eQTL target variance can be explained by control at the level of transcription, but that the remaining 25% may be regulated co- or post-transcriptionally. We focus on eQTL targets with discordant mRNA and pre-mRNA expression patterns and use four examples: USMG5, MMAB, MRPL43, and OAS1, to dissect the exact downstream effects of the associated genetic variants.
Figures
References
-
- Andrés AM, Dennis MY, Kretzschmar WW, Cannons JL, Lee-Lin SQ, Hurle B, NISC Comparative Sequencing Program, Schwartzberg PL, Williamson SH, Bustamante CD, et al. 2010. Balancing selection maintains a form of ERAP2 that undergoes nonsense-mediated decay and affects antigen presentation. PLoS Genet 6: e1001157 doi: 10.1371/journal.pgen.1001157 - PMC - PubMed
-
- Bonnevie-Nielsen V, Field LL, Lu S, Zheng DJ, Li M, Martensen PM, Nielsen TB, Beck-Nielsen H, Lau YL, Pociot F 2005. Variation in antiviral 2′,5′-oligoadenylate synthetase (2′5′AS) enzyme activity is controlled by a single-nucleotide polymorphism at a splice-acceptor site in the OAS1 gene. Am J Hum Genet 76: 623–633 - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials