

A novel epigenetic phenotype associated with the most aggressive pathway of bladder tumor progression
- PMID: 21173382
- PMCID: PMC3014990
- DOI: 10.1093/jnci/djq470
A novel epigenetic phenotype associated with the most aggressive pathway of bladder tumor progression
Abstract
Background: Epigenetic silencing can extend to whole chromosomal regions in cancer. There have been few genome-wide studies exploring its involvement in tumorigenesis.
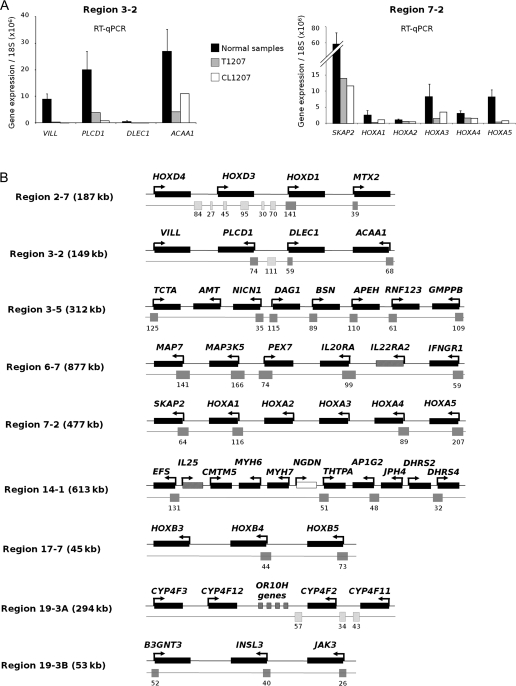
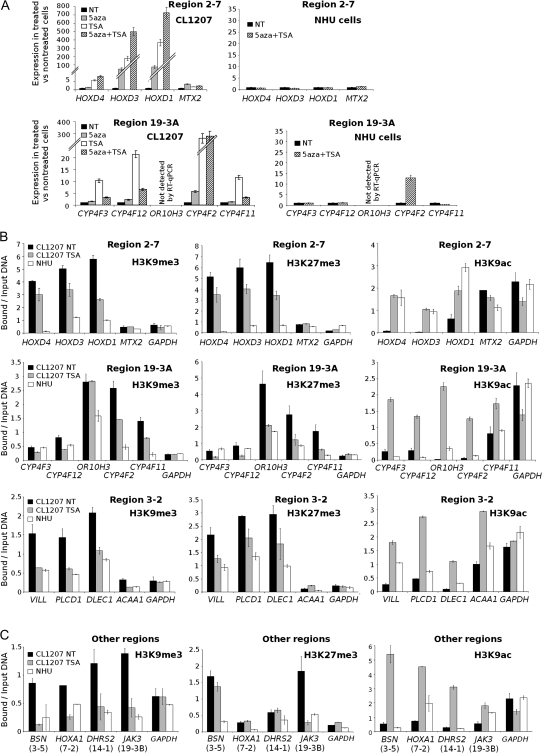
Methods: We searched for chromosomal regions affected by epigenetic silencing in cancer by using Affymetrix microarrays and real-time quantitative polymerase chain reaction to analyze RNA from 57 bladder tumors compared with normal urothelium. Epigenetic silencing was verified by gene re-expression following treatment of bladder cell lines with 5-aza-deoxycytidine, a DNA demethylating agent, and trichostatin A, a histone deacetylase inhibitor. DNA methylation was studied by bisulfite sequencing and histone methylation and acetylation by chromatin immunoprecipitation. Clustering was used to distinguish tumors with multiple regional epigenetic silencing (MRES) from those without and to analyze the association of this phenotype with histopathologic and molecular types of bladder cancer. The results were confirmed with a second panel of 40 tumor samples and extended in vitro with seven bladder cancer cell lines. All statistical tests were two-sided.
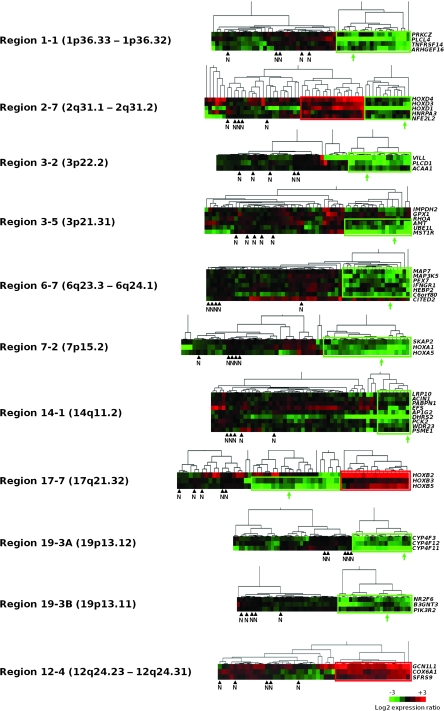
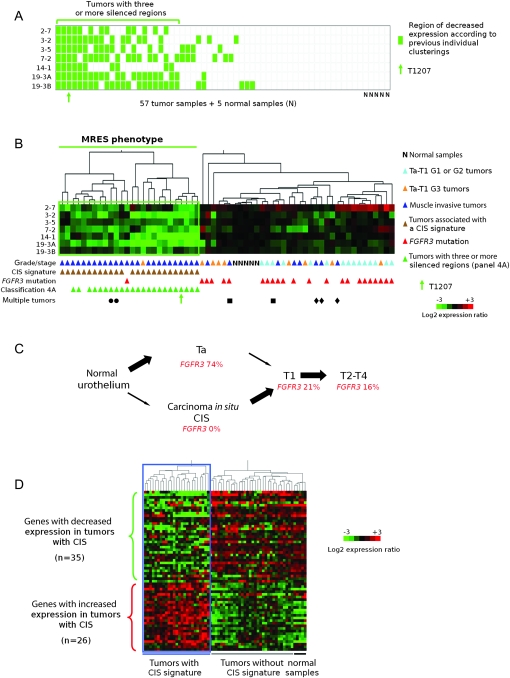
Results: We identified seven chromosomal regions of contiguous genes that were silenced by an epigenetic mechanism. Epigenetic silencing was not associated with DNA methylation but was associated with histone H3K9 and H3K27 methylation and histone H3K9 hypoacetylation. All seven regions were concordantly silenced in a subgroup of 26 tumors, defining an MRES phenotype. MRES tumors exhibited a carcinoma in situ-associated gene expression signature (25 of 26 MRES tumors vs 0 of 31 non-MRES tumors, P < 10⁻¹⁴), rarely carried FGFR3 mutations (one of 26 vs 22 of 31 non-MRES tumors, P < 10⁻¹⁶), and contained 25 of 33 (76%) of the muscle-invasive tumors. Cell lines derived from aggressive bladder tumors presented epigenetic silencing of the same regions.
Conclusions: We have identified an MRES phenotype characterized by the concomitant epigenetic silencing of several chromosomal regions, which, in bladder cancer, is specifically associated with the carcinoma in situ gene expression signature.
Figures

References
-
- Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3(6):415–428. - PubMed
-
- Laird PW. Cancer epigenetics. Hum Mol Genet. 2005;14(Spec No 1):R65–R76. - PubMed
-
- Esteller M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet. 2007;8(4):286–298. - PubMed
-
- Gibbons RJ. Histone modifying and chromatin remodelling enzymes in cancer and dysplastic syndromes. Hum Mol Genet. 2005;14(Spec No 1):R85–R92. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
