

Hearing loss: a common disorder caused by many rare alleles
- PMID: 21175685
- PMCID: PMC3689008
- DOI: 10.1111/j.1749-6632.2010.05868.x
Hearing loss: a common disorder caused by many rare alleles
Abstract
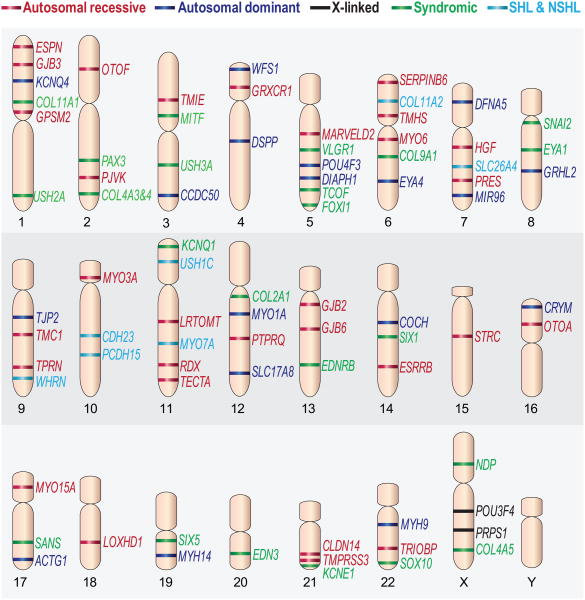
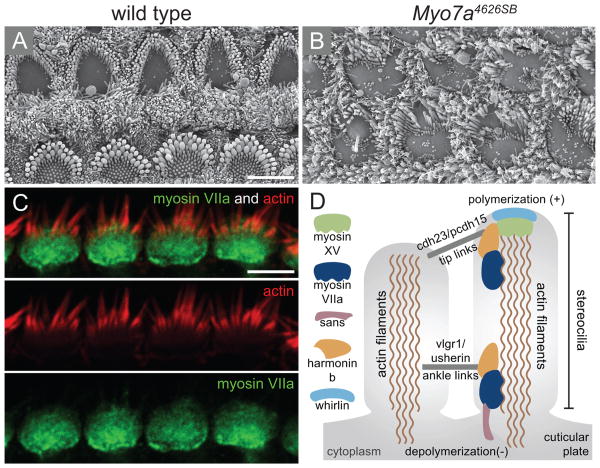
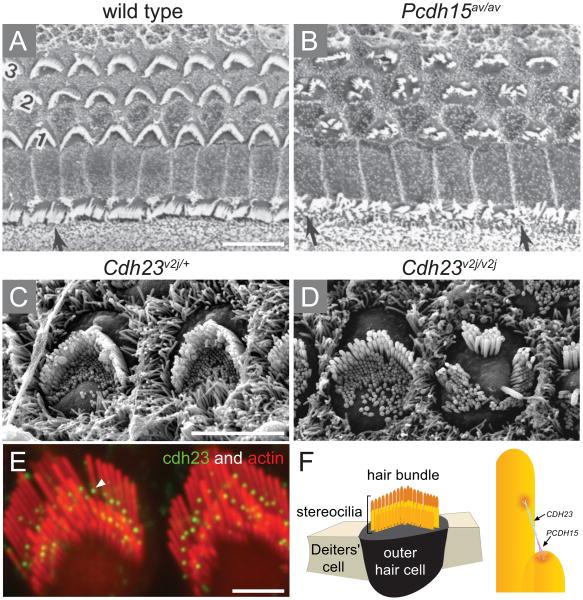
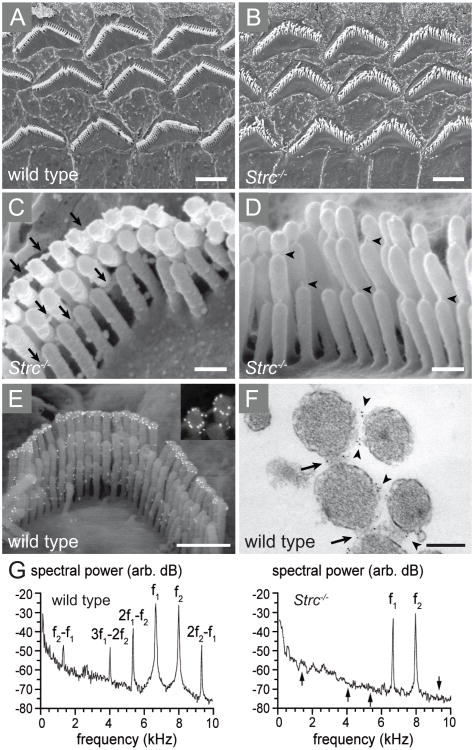
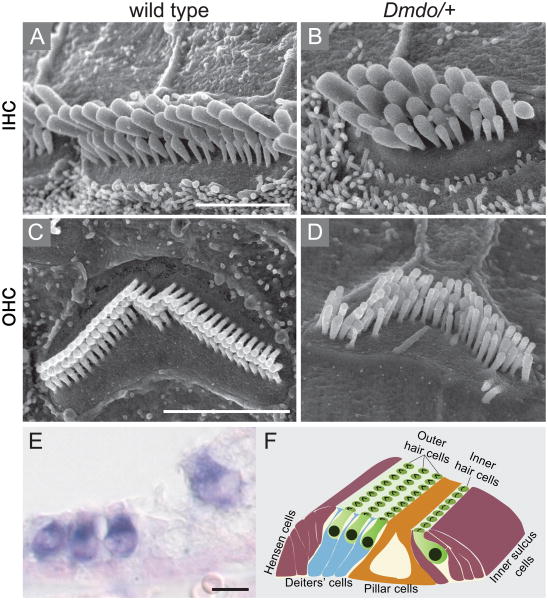
Perception of sound is a fundamental role of the auditory system. Traveling with the force of their mechanical energy, sound waves are captured by the ear and activate the sensory pathway of this complex organ. The hair cells, specialized sensory cells within the inner ear, transmit the mechanical energy into electrical nerve stimuli that reach the brain. A large number of proteins are responsible for the overarching tasks required to maintain the complex mechanism of sound sensation. Many hearing disorders are due to single gene defects inherited in a Mendelian fashion, thus enabling clinical diagnostics. However, at the same time, hearing impairment is genetically heterogeneous, with both common and rare forms occurring due to mutations in over 100 genes. The crosstalk between human and mouse genetics has enabled comprehensive studies on gene identification and protein function, taking advantage of the tools animal models have to offer. The aim of the following review is to provide background and examples of human deafness genes and the discovery of their function in the auditory system.
© 2010 New York Academy of Sciences.
Figures
References
-
- Petit C, Levilliers J, Hardelin JP. Molecular genetics of hearing loss. Annu Rev Genet. 2001;35:589–646. - PubMed
-
- Gorlin RJ. Genetic Hearing Loss-A Brief History. In: Gorlin RJ, Toriello HV, Cohen MM, editors. Hereditary Hearing Loss and its Syndromes. Oxford University Press; New York: 1995. pp. 3–4.
-
- Dow GS, Poynter CI. The Dar family. Eugen News. 1930;15:128–130.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
