

seqMINER: an integrated ChIP-seq data interpretation platform
- PMID: 21177645
- PMCID: PMC3064796
- DOI: 10.1093/nar/gkq1287
seqMINER: an integrated ChIP-seq data interpretation platform
Abstract
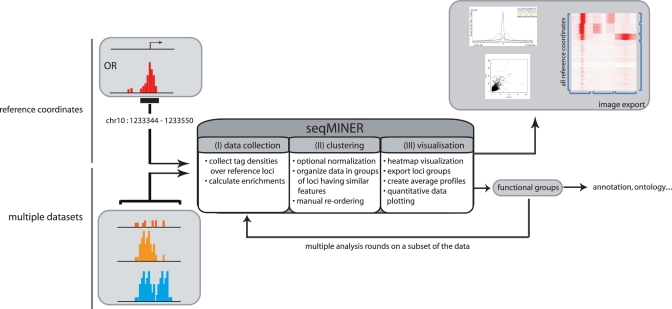
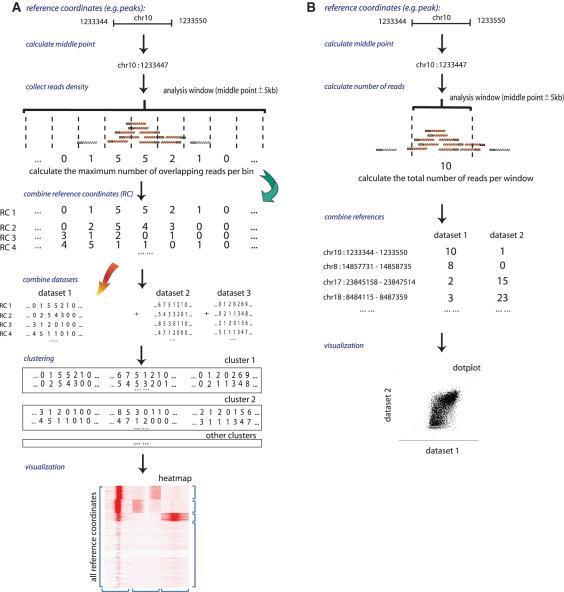
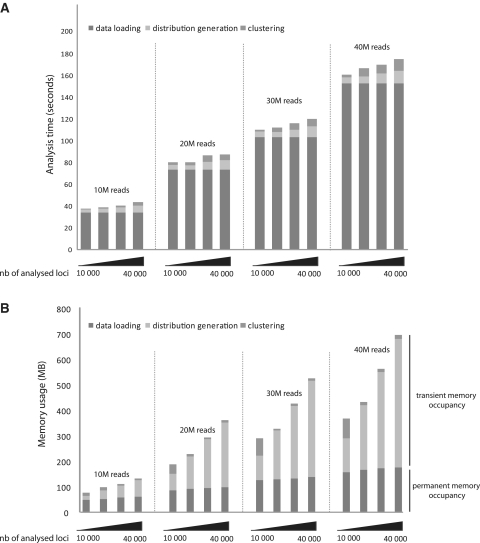
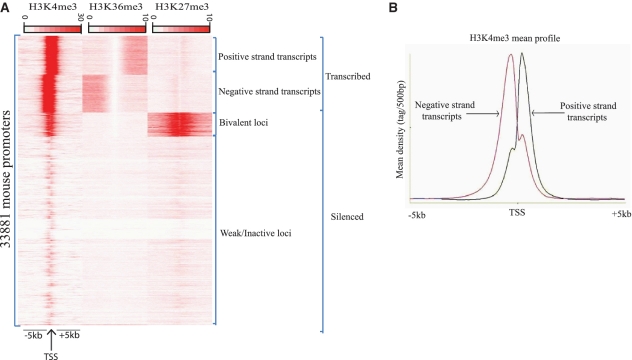
In a single experiment, chromatin immunoprecipitation combined with high throughput sequencing (ChIP-seq) provides genome-wide information about a given covalent histone modification or transcription factor occupancy. However, time efficient bioinformatics resources for extracting biological meaning out of these gigabyte-scale datasets are often a limiting factor for data interpretation by biologists. We created an integrated portable ChIP-seq data interpretation platform called seqMINER, with optimized performances for efficient handling of multiple genome-wide datasets. seqMINER allows comparison and integration of multiple ChIP-seq datasets and extraction of qualitative as well as quantitative information. seqMINER can handle the biological complexity of most experimental situations and proposes methods to the user for data classification according to the analysed features. In addition, through multiple graphical representations, seqMINER allows visualization and modelling of general as well as specific patterns in a given dataset. To demonstrate the efficiency of seqMINER, we have carried out a comprehensive analysis of genome-wide chromatin modification data in mouse embryonic stem cells to understand the global epigenetic landscape and its change through cellular differentiation.
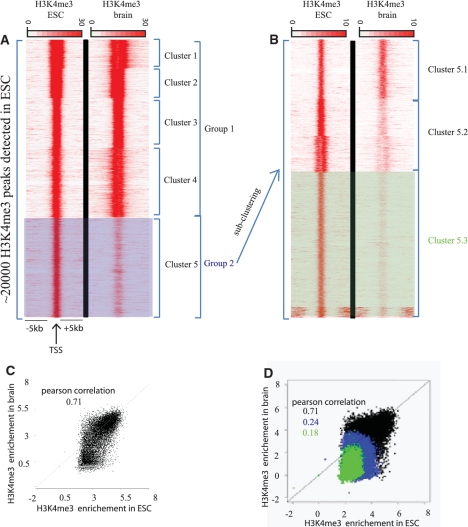
Figures
References
-
- Johnson DS, Mortazavi A, Myers RM, Wold B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;316:1497–1502. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
